2026
-
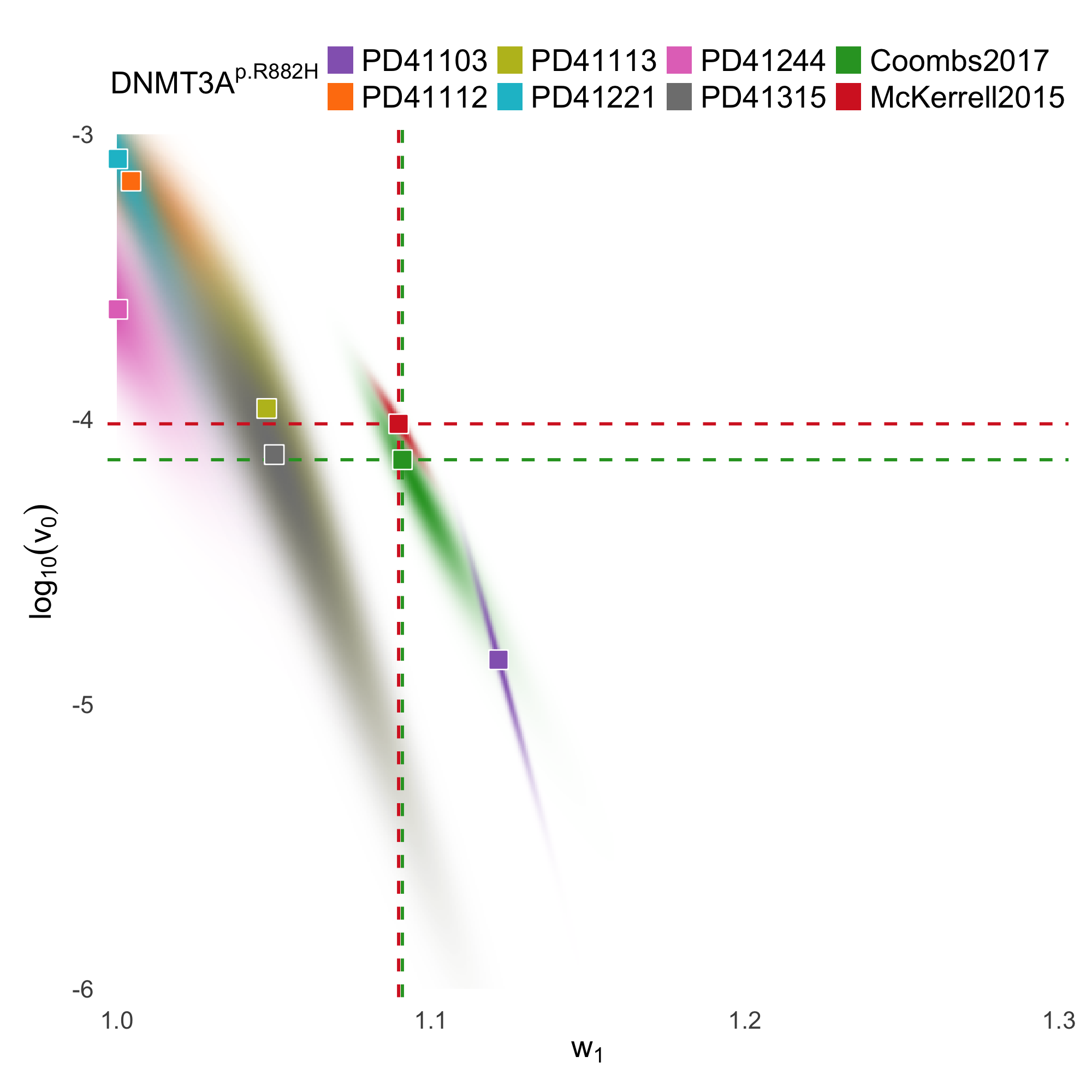 BESTish: A diffusion-approximation framework for inferring selection and mutation in clonal hematopoiesisbioRxiv, 2026
BESTish: A diffusion-approximation framework for inferring selection and mutation in clonal hematopoiesisbioRxiv, 2026Clonal hematopoiesis (CH) arises when hematopoietic stem cells (HSCs) gain a fitness advantage from somatic mutations and expand, resulting in an increase in variant allele frequency (VAF) over time. To analyze CH trajectories, we develop a state-dependent stochastic model of wild-type and mutant HSCs, in which an environmental parameter α∈[0,1] regulates death rates and interpolates between homeostatic (Moran-like, α=1) and growth-facilitating (α<1) regimes. Using functional law of large numbers and central limit theorems, we derive explicit mean-field dynamics and a Gaussian-Markov approximation for VAF fluctuations. We show that the mean VAF trajectory has an explicit logistic form determined by selective advantage, while environmental effects affect only the variance and autocovariance structure. Building on these results, we introduce BESTish (Bayesian estimate for selection incorporating scaling-limit to detect mutant heterogeneity), a novel, efficient and accurate Bayesian inference method that can be applied to analyze both cohort-level and longitudinal VAF datasets. BESTish implements the closed-form finite-dimensional distributions that we derive to estimate mutation fitness, mutation rate, and environmental strength for individual CH drivers. When applied to existing CH datasets, BESTish produces consistent mutation fitness inferences across different studies, and estimates CH driver mutation rates in agreement with independent experimental studies. Furthermore, BESTish reveals patient-specific heterogeneity in the selective behavior of recurrent mutations, and identifies variants whose dynamics are compatible with non-homeostatic, growth-facilitating environments. BESTish provides a unified and mechanistic framework for quantifying CH evolution, with potential applications for other biological systems where clonal expansions can be measured.
@article{wang2026bestish, dimensions = {true}, title = {BESTish: A diffusion-approximation framework for inferring selection and mutation in clonal hematopoiesis}, author = {Wang, Ren-Yi and Dinh, Khanh N. and Taketomi, Keito and Pang, Guodong and King, Katherine Y. and Kimmel, Marek}, journal = {bioRxiv}, year = {2026}, doi = {10.64898/2026.01.27.702030}, } -
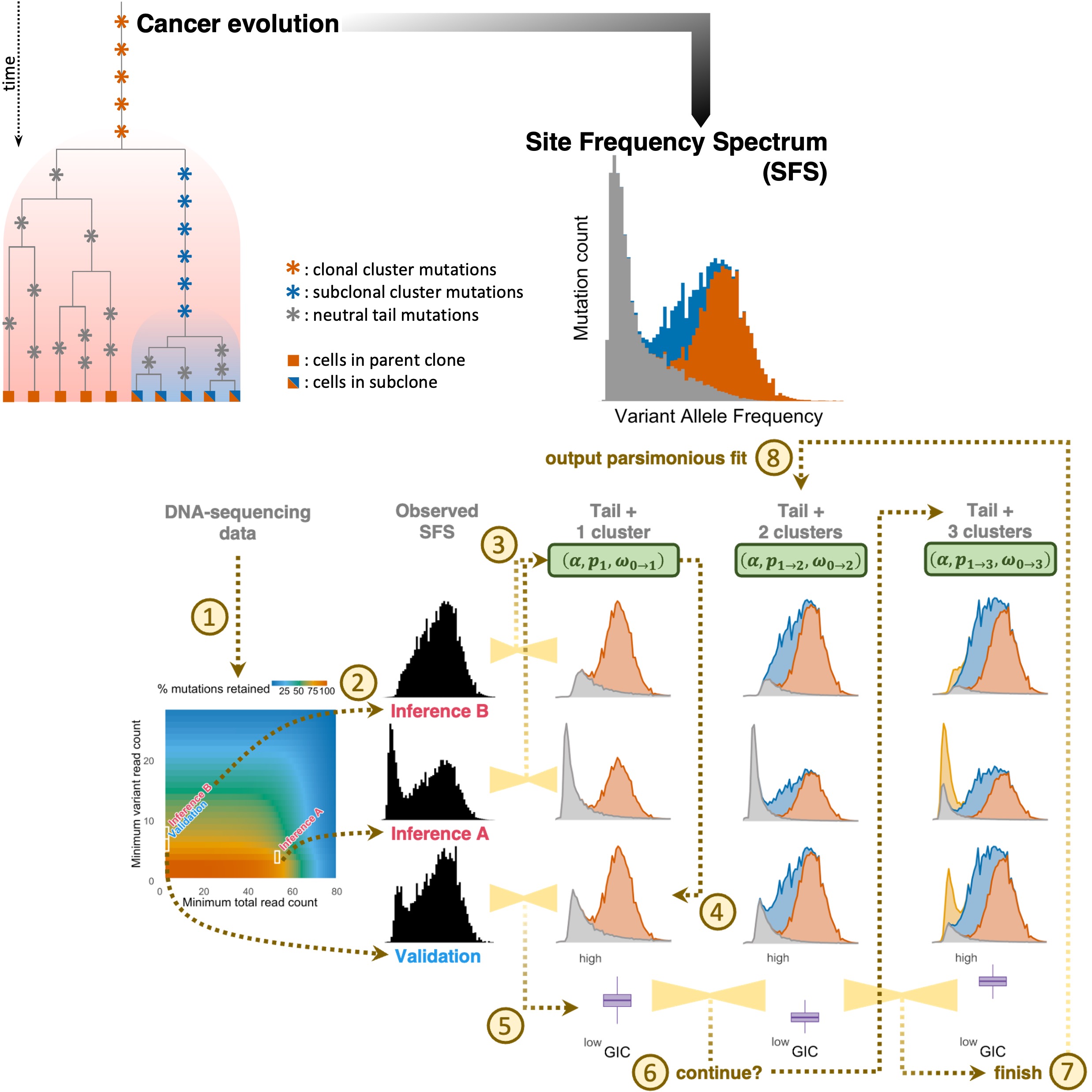 Accurate detection of tumor clonality and ongoing expansion mode from genomic dataYanjie Chen, Roman Jaksik, Peter Terranova, Sara El Baghdadi, Andrew Koval, Monika K. Kurpas, Simon Tavaré, and Khanh N. DinhbioRxiv, 2026
Accurate detection of tumor clonality and ongoing expansion mode from genomic dataYanjie Chen, Roman Jaksik, Peter Terranova, Sara El Baghdadi, Andrew Koval, Monika K. Kurpas, Simon Tavaré, and Khanh N. DinhbioRxiv, 2026Recent evidence shows that despite considerable effort, currently available algorithms for estimating intra-tumor heterogeneity (ITH) remain limited. We developed DECODE (Deciphering Cancer Origin from DNA Evolution), a novel mutation clustering method that incorporates the impact of sample-specific sequencing coverage and mutation calling biases. On synthetic data, DECODE outperformed existing methods across multiple clonality metrics and accurately detected and characterized the neutral tail in the site frequency spectrum (SFS), which encodes the tumor’s ongoing expansion mode. In acute myeloid leukemia, accounting for the neutral tail enabled DECODE to yield more parsimonious clonal decompositions that align more closely with known subclonal dynamics that drive relapse. Applied to data from The Cancer Genome Atlas, DECODE not only detected a neutral SFS tail in most samples across tumor types but also uncovered a clinically meaningful link between ITH and survival in low-grade glioma. By jointly inferring clonality and expansion mode, DECODE provides two complementary and prognostically relevant readouts of tumor evolution from single tumor genomic samples.
@article{chen2026accurate, dimensions = {true}, title = {Accurate detection of tumor clonality and ongoing expansion mode from genomic data}, author = {Chen, Yanjie and Jaksik, Roman and Terranova, Peter and El Baghdadi, Sara and Koval, Andrew and Kurpas, Monika K. and Tavar{\'e}, Simon and Dinh, Khanh N.}, journal = {bioRxiv}, year = {2026}, doi = {10.64898/2026.06.15.732415}, } -
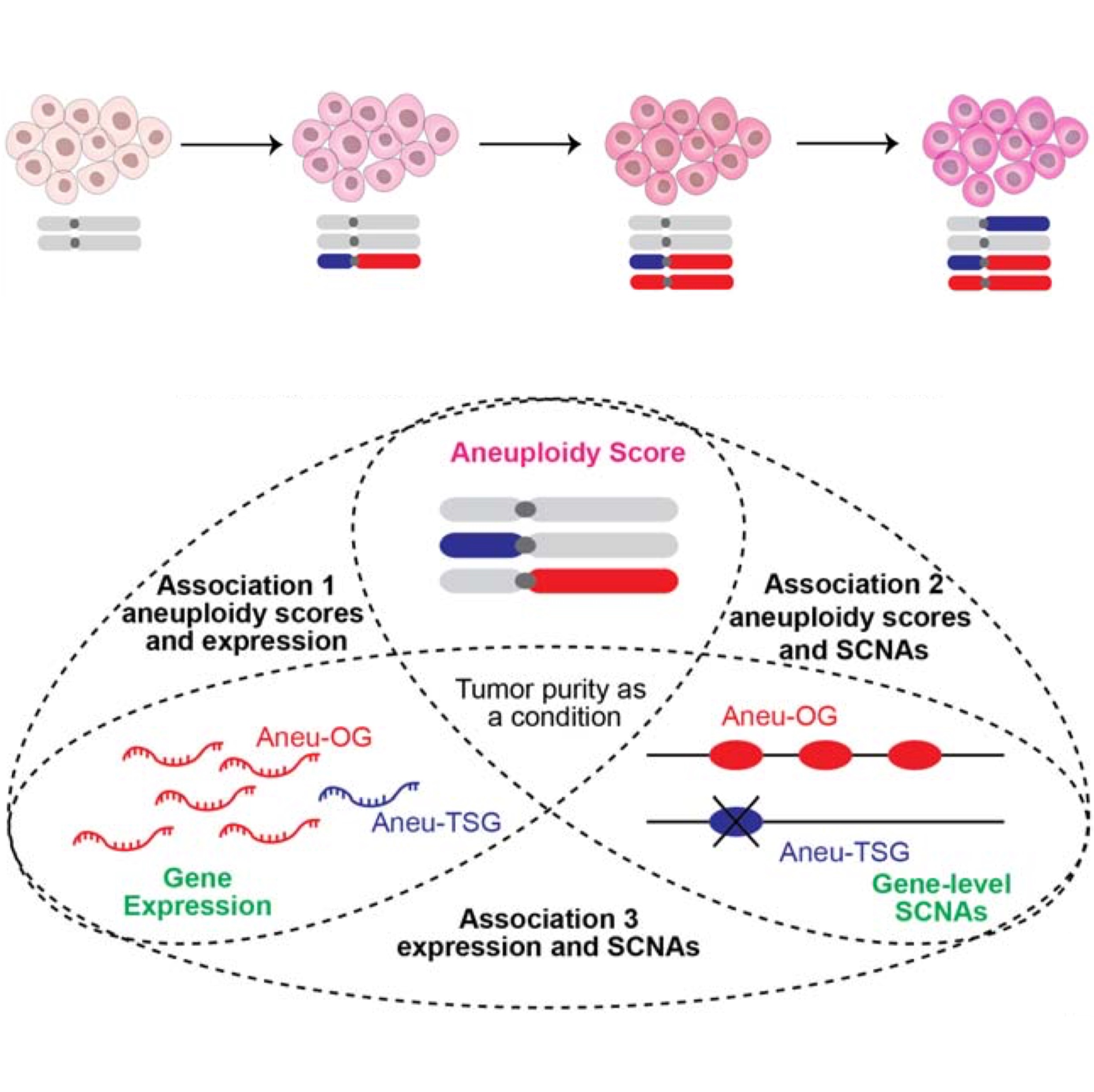 COATS identifies copy-number-dependent drivers and enablers of aneuploidy in cancerGaojianyong Wang, Khanh N. Dinh, Fabio Alfieri, Somayeh Fani, and Teresa DavolibioRxiv, 2026
COATS identifies copy-number-dependent drivers and enablers of aneuploidy in cancerGaojianyong Wang, Khanh N. Dinh, Fabio Alfieri, Somayeh Fani, and Teresa DavolibioRxiv, 2026Aneuploidy is a hallmark of most cancers but at the same time has been shown to decrease cellular fitness. Thus, to solve this conundrum, a current hypothesis in the field is that specific SCNAs may promote tolerance to the aneuploid state and/or promote additional chromosomal instability (CIN) and more aneuploidy. In other words, gains or losses of oncogenes (OGs) and tumor suppressor genes (TSGs) can, in turn, drive further CIN, promoting additional somatic alterations, or enhance aneuploid cell survival. Despite their importance, CN-dependent OGs and TSGs associated with aneuploidy remain largely unidentified. Here, we present a new method, Copy-number-dependent Oncogenes And Tumor Suppressors (COATS), to identify pan-cancer and cancer-specific CN-dependent OGs and TSGs associated with aneuploidy (Aneu-OGs and Aneu-TSGs). COATS integrates information theory and statistical tests to analyze gene expression, copy number, and aneuploidy, and incorporates timing analysis to distinguish early drivers of CIN from late tolerance enablers. Interestingly, using the CINner simulation framework, we show that aneuploidy drivers tend to occur earlier than aneuploidy enablers. Applying COATS to 33 TCGA cancer types, we identified 479 pan-cancer amplification-dependent Aneu-OGs and 141 deletion-dependent Aneu-TSGs. For validation, we used shRNA to knock down CCT5, a COATS-identified pan-cancer Aneu-OG predicted to promote aneuploidy tolerance, in isogenic aneuploid and near-diploid cells. Strikingly, CCT5 depletion was selectively toxic in aneuploid cells, supporting its classification as an aneuploidy tolerance enabler gene. Overall, our study defines a set of CN-dependent genes associated with aneuploidy and points to candidate therapeutic targets for chromosomally unstable cancers.
@article{wang2026coats, dimensions = {true}, title = {COATS identifies copy-number-dependent drivers and enablers of aneuploidy in cancer}, author = {Wang, Gaojianyong and Dinh, Khanh N. and Alfieri, Fabio and Fani, Somayeh and Davoli, Teresa}, journal = {bioRxiv}, year = {2026}, doi = {10.64898/2026.06.22.733880}, }
2025
-
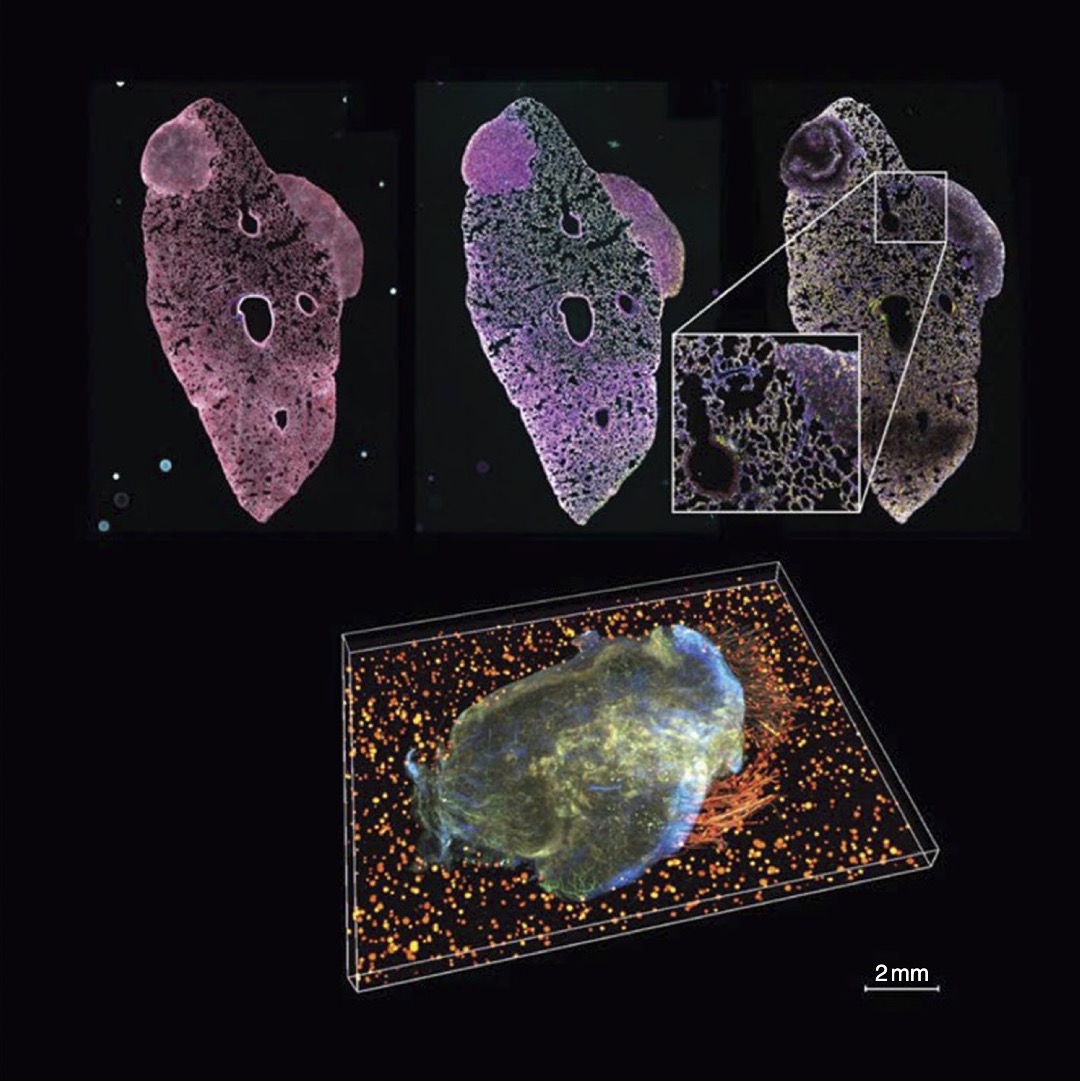 Cancer research in the age of spatial omics: lessons from IMAXTDario Bressan, IMAXT Consortium, Nicholas Walton, and Gregory J. HannonCancer Discovery, 2025
Cancer research in the age of spatial omics: lessons from IMAXTDario Bressan, IMAXT Consortium, Nicholas Walton, and Gregory J. HannonCancer Discovery, 2025The Imaging and Molecular Annotation of Xenografts and Tumors Cancer Grand Challenges team was set up with the objective of developing the “next generation” of pathology and cancer research by using a combination of single-cell and spatial omics tools to produce 3D molecularly annotated maps of tumors. Its activities overlapped, and in some cases catalyzed, a spatial revolution in biology that saw new technologies being deployed to investigate the roles of tumor heterogeneity and of the tumor micro-environment.
@article{bressan2025cancer, dimensions = {true}, title = {Cancer research in the age of spatial omics: lessons from IMAXT}, author = {Bressan, Dario and Consortium, IMAXT and Walton, Nicholas and Hannon, Gregory J.}, journal = {Cancer Discovery}, volume = {15}, number = {1}, pages = {16--21}, year = {2025}, publisher = {American Association for Cancer Research}, doi = {10.1158/2159-8290.CD-24-1686}, } -
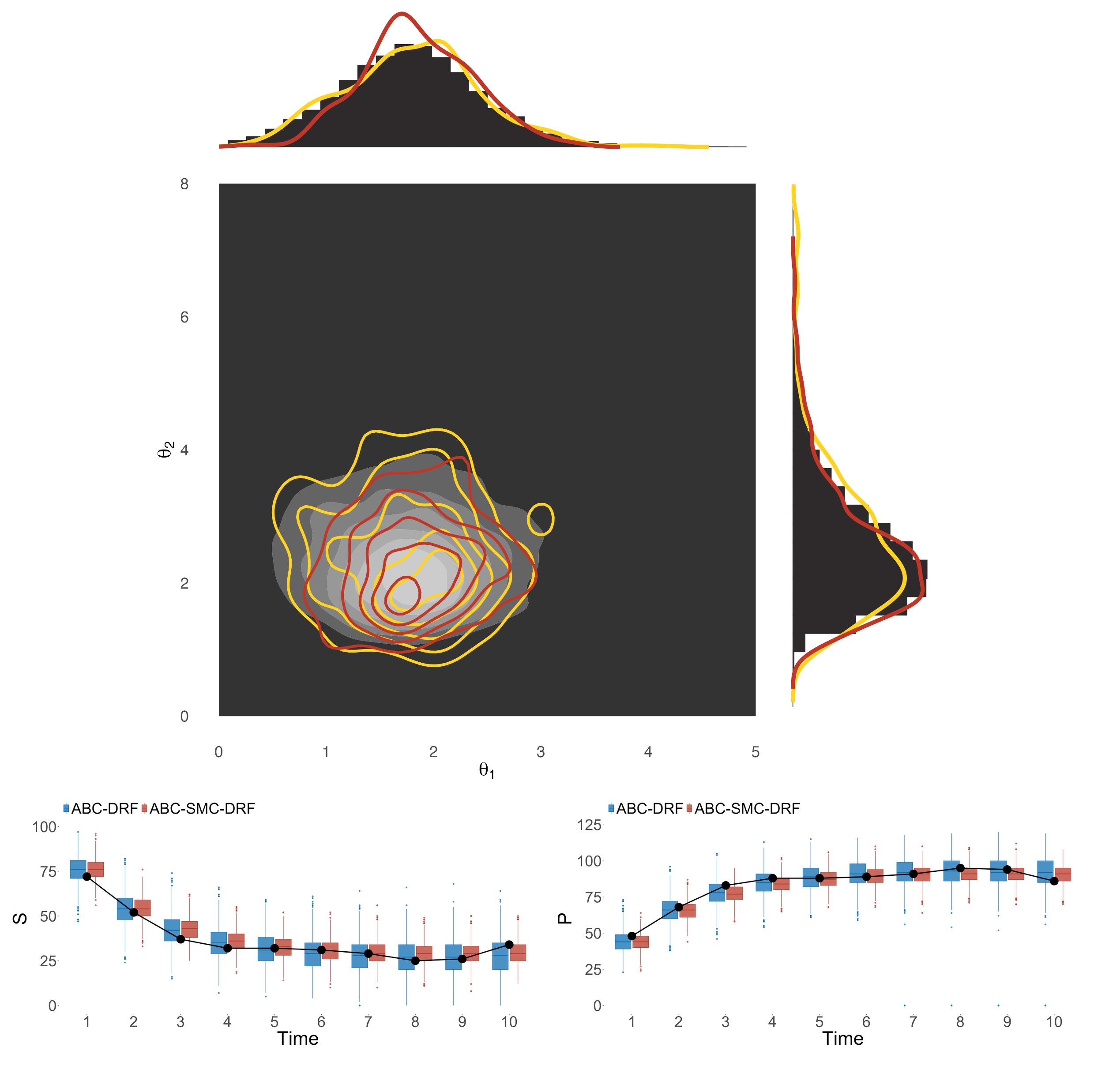 Approximate Bayesian computation sequential Monte Carlo via random forestsStatistics and Computing, 2025
Approximate Bayesian computation sequential Monte Carlo via random forestsStatistics and Computing, 2025Approximate Bayesian Computation (ABC) is a popular inference method when likelihoods are hard to come by. Practical bottlenecks of ABC applications include selecting statistics that summarize the data without losing too much information or introducing uncertainty, and choosing distance functions and tolerance thresholds that balance accuracy and computational efficiency. Recent studies have shown that ABC methods using random forest (RF) methodology perform well while circumventing many of ABC’s drawbacks. However, RF construction is computationally expensive for large numbers of trees and model simulations, and there can be high uncertainty in the posterior if the prior distribution is uninformative. Here we further adapt random forests to the ABC setting in two ways. The first exploits distributional random forests to provide a direct method for inferring the joint posterior distribution of parameters of interest, while the second describes a sequential Monte Carlo approach which updates the prior distribution iteratively to focus on the most likely regions in the parameter space. We show that the new methods can accurately infer posterior distributions for a wide range of deterministic and stochastic models in different scientific areas.
@article{dinh2025approximate, dimensions = {true}, title = {Approximate Bayesian computation sequential Monte Carlo via random forests}, author = {Dinh, Khanh N. and Liu, C{\'e}cile and Xiang, Zijin and Liu, Zhihan and Tavar{\'e}, Simon}, journal = {Statistics and Computing}, year = {2025}, doi = {10.1007/s11222-025-10748-x}, } -
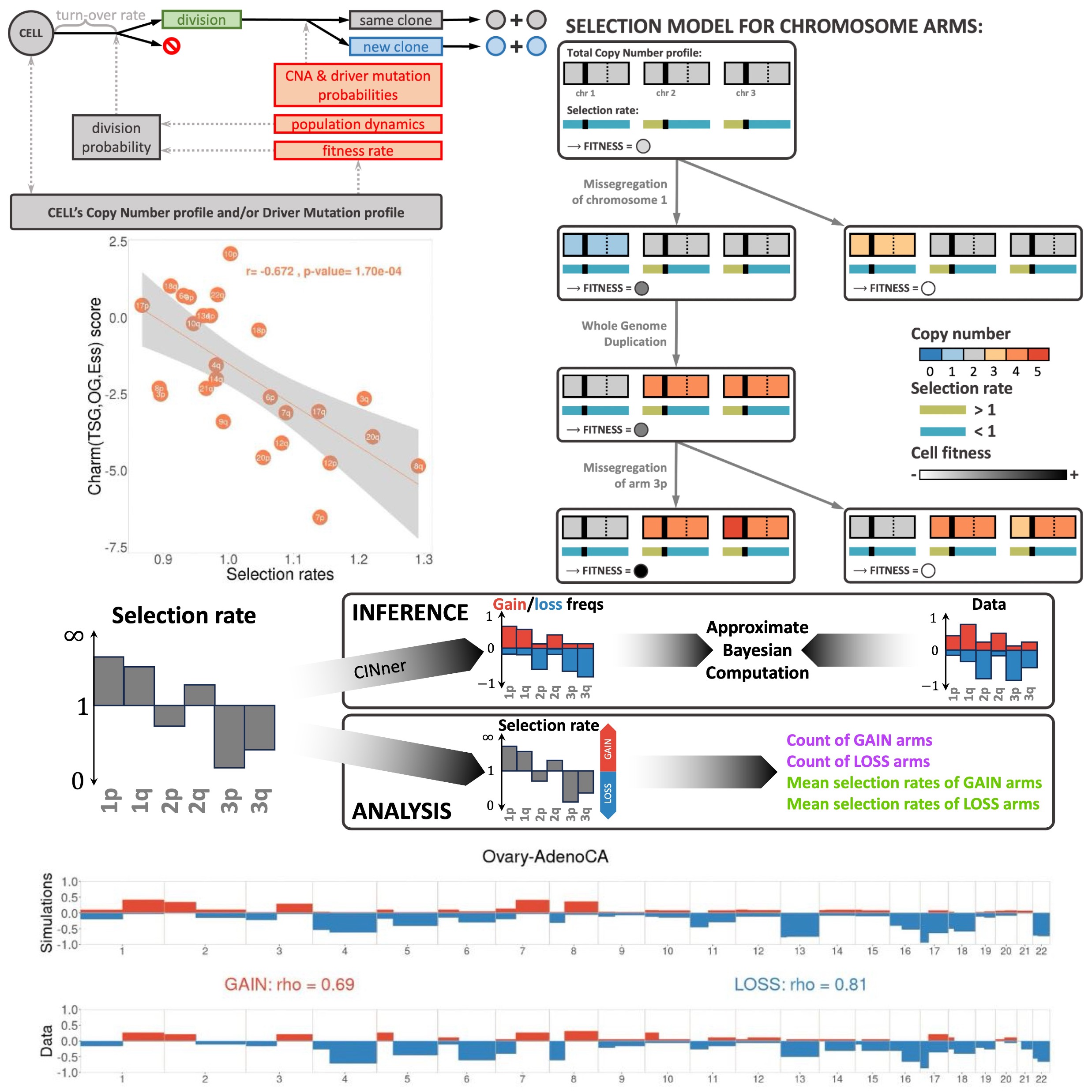 CINner: modeling and simulation of chromosomal instability in cancer at single-cell resolutionKhanh N. Dinh, Ignacio Vázquez-García, Andrew Chan, Rhea Malhotra, Adam Weiner, Andrew W. McPherson, and Simon TavaréPLoS Computational Biology, 2025
CINner: modeling and simulation of chromosomal instability in cancer at single-cell resolutionKhanh N. Dinh, Ignacio Vázquez-García, Andrew Chan, Rhea Malhotra, Adam Weiner, Andrew W. McPherson, and Simon TavaréPLoS Computational Biology, 2025Cancer development is characterized by chromosomal instability, manifesting in frequent occurrences of different genomic alteration mechanisms ranging in extent and impact. Mathematical modeling can help evaluate the role of each mutational process during tumor progression, however existing frameworks can only capture certain aspects of chromosomal instability (CIN). We present CINner, a mathematical framework for modeling genomic diversity and selection during tumor evolution. The main advantage of CINner is its flexibility to incorporate many genomic events that directly impact cellular fitness, from driver gene mutations to copy number alterations (CNAs), including focal amplifications and deletions, missegregations and whole-genome duplication (WGD). We apply CINner to find chromosome-arm selection parameters that drive tumorigenesis in the absence of WGD in chromosomally unstable cancer types from the Pan-Cancer Analysis of Whole Genomes (PCAWG, n=718). We found that the selection parameters predict WGD prevalence among different chromosomally unstable tumors, hinting that the selective advantage of WGD cells hinges on their tolerance for aneuploidy and escape from nullisomy. Analysis of inference results using CINner across cancer types in The Cancer Genome Atlas (n=8207) further reveals that the inferred selection parameters reflect the bias between tumor suppressor genes and oncogenes on specific genomic regions. Direct application of CINner to model the WGD proportion and fraction of genome altered (FGA) in PCAWG uncovers the increase in CNA probabilities associated with WGD in each cancer type. CINner can also be utilized to study chromosomally stable cancer types, by applying a selection model based on driver gene mutations and focal amplifications or deletions (chronic lymphocytic leukemia in PCAWG, n=95). Finally, we used CINner to analyze the impact of CNA probabilities, chromosome selection parameters, tumor growth dynamics and population size on cancer fitness and heterogeneity. We expect that CINner will provide a powerful modeling tool for the oncology community to quantify the impact of newly uncovered genomic alteration mechanisms on shaping tumor progression and adaptation.
@article{dinh2025cinner, dimensions = {true}, title = {CINner: modeling and simulation of chromosomal instability in cancer at single-cell resolution}, author = {Dinh, Khanh N. and Vázquez-García, Ignacio and Chan, Andrew and Malhotra, Rhea and Weiner, Adam and McPherson, Andrew W. and Tavar{\'e}, Simon}, journal = {PLoS Computational Biology}, year = {2025}, doi = {10.1371/journal.pcbi.1012902}, } -
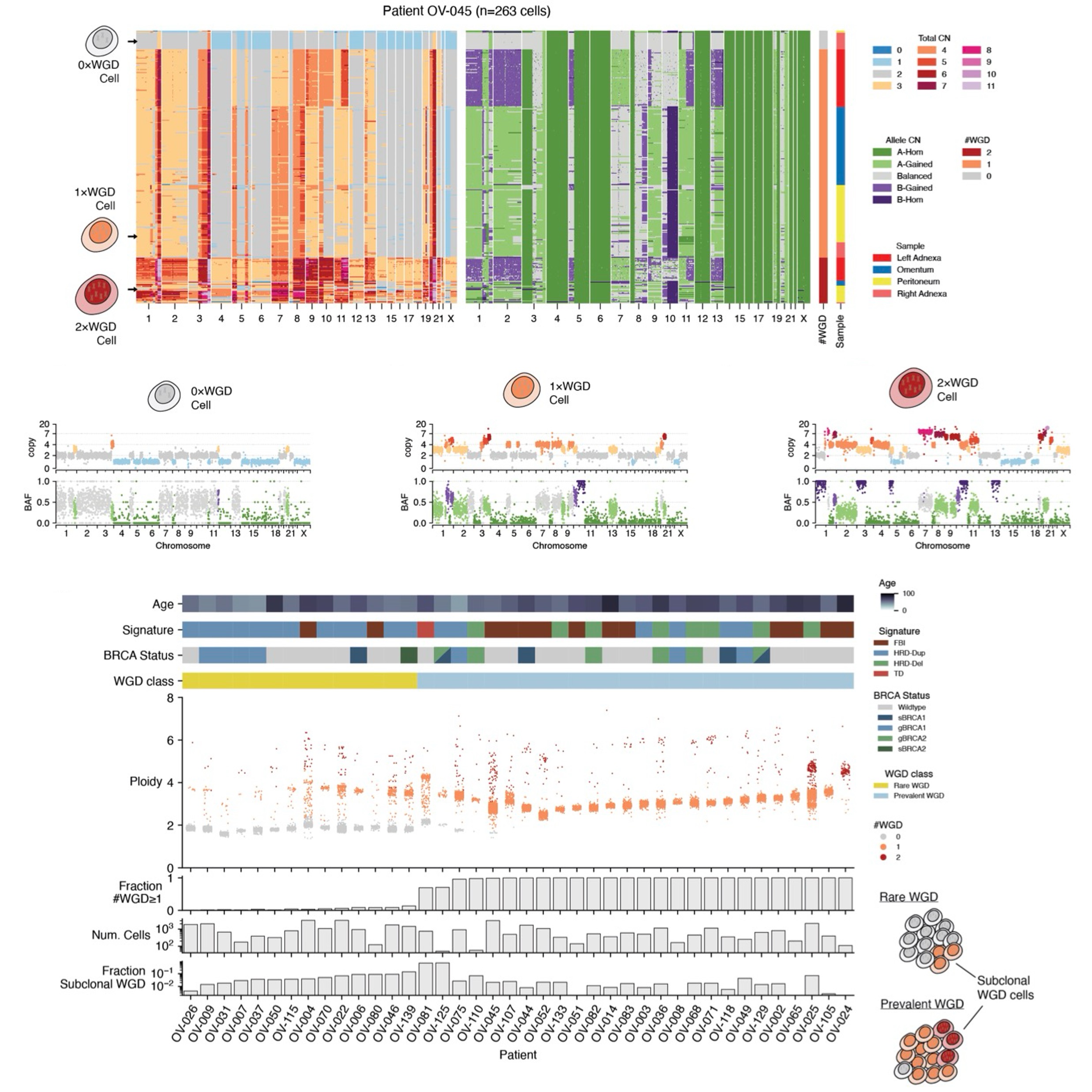 Ongoing genome doubling shapes evolvability and immunity in ovarian cancerAndrew W. McPherson, Ignacio Vázquez-García, Matthew A. Myers, Duaa H. Al-Rawi, Matthew Zatzman, Adam C. Weiner, Samuel S. Freeman, Neeman Mohibullah, Gryte Satas, Marc J. Williams, Nicholas Ceglia, Danguolė Norbūnaitė, Allen W. Zhang, Jun Li, Jamie L. P. Lim, Michele Wu, Seongmin Choi, Eliyahu Havasov, Diljot Grewal, Hongyu Shi, Minsoo Kim, Roland F. Schwarz, Tom Kaufmann, Khanh N. Dinh, Florian Uhlitz, Julie Tran, Yushi Wu, Ruchi Patel, Satish Ramakrishnan, DooA Kim, Justin Clarke, Hunter Green, Emily Ali, Melody DiBona, Nancy Varice, Ritika Kundra, Vance Broach, Ginger J. Gardner, Kara L. Roche, Yukio Sonoda, Oliver Zivanovic, Sarah H. Kim, Rachel N. Grisham , Ying L. Liu, Agnes Viale, Nicole Rusk, Yulia Lakhman, Lora H. Ellenson, Simon Tavaré, Samuel Aparicio, Dennis S. Chi, Carol Aghajanian, Nadeem R. Abu-Rustum, Claire F. Friedman, Dmitriy Zamarin, Britta Weigelt, Samuel F. Bakhoum, and Sohrab P. ShahNature, 2025
Ongoing genome doubling shapes evolvability and immunity in ovarian cancerAndrew W. McPherson, Ignacio Vázquez-García, Matthew A. Myers, Duaa H. Al-Rawi, Matthew Zatzman, Adam C. Weiner, Samuel S. Freeman, Neeman Mohibullah, Gryte Satas, Marc J. Williams, Nicholas Ceglia, Danguolė Norbūnaitė, Allen W. Zhang, Jun Li, Jamie L. P. Lim, Michele Wu, Seongmin Choi, Eliyahu Havasov, Diljot Grewal, Hongyu Shi, Minsoo Kim, Roland F. Schwarz, Tom Kaufmann, Khanh N. Dinh, Florian Uhlitz, Julie Tran, Yushi Wu, Ruchi Patel, Satish Ramakrishnan, DooA Kim, Justin Clarke, Hunter Green, Emily Ali, Melody DiBona, Nancy Varice, Ritika Kundra, Vance Broach, Ginger J. Gardner, Kara L. Roche, Yukio Sonoda, Oliver Zivanovic, Sarah H. Kim, Rachel N. Grisham , Ying L. Liu, Agnes Viale, Nicole Rusk, Yulia Lakhman, Lora H. Ellenson, Simon Tavaré, Samuel Aparicio, Dennis S. Chi, Carol Aghajanian, Nadeem R. Abu-Rustum, Claire F. Friedman, Dmitriy Zamarin, Britta Weigelt, Samuel F. Bakhoum, and Sohrab P. ShahNature, 2025Whole-genome doubling (WGD) is a common feature of human cancers and is linked to tumour progression, drug resistance, and metastasis. Here we examine the impact of WGD on somatic evolution and immune evasion at single-cell resolution in patient tumours. Using single-cell whole-genome sequencing, we analysed 70 high-grade serous ovarian cancer samples from 41 patients (30,260 tumour genomes) and observed near-ubiquitous evidence that WGD is an ongoing mutational process. WGD was associated with increased cell-cell diversity and higher rates of chromosomal missegregation and consequent micronucleation. We developed a mutation-based WGD timing method called doubleTime to delineate specific modes by which WGD can drive tumour evolution, including early fixation followed by considerable diversification, multiple parallel WGD events on a pre-existing background of copy-number diversity, and evolutionarily late WGD in small clones and individual cells. Furthermore, using matched single-cell RNA sequencing and high-resolution immunofluorescence microscopy, we found that inflammatory signalling and cGAS-STING pathway activation result from ongoing chromosomal instability, but this is restricted to predominantly diploid tumours (WGD-low). By contrast, predominantly WGD tumours (WGD-high), despite increased missegregation, exhibited cell-cycle dysregulation, STING1 repression, and immunosuppressive phenotypic states. Together, these findings establish WGD as an ongoing mutational process that promotes evolvability and dysregulated immunity in high-grade serous ovarian cancer.
@article{mcpherson2025ongoing, dimensions = {true}, title = {Ongoing genome doubling shapes evolvability and immunity in ovarian cancer}, author = {McPherson, Andrew W. and Vázquez-García, Ignacio and Myers, Matthew A. and Al-Rawi, Duaa H. and Zatzman, Matthew and Weiner, Adam C. and Freeman, Samuel S. and Mohibullah, Neeman and Satas, Gryte and Williams, Marc J. and Ceglia, Nicholas and Norb\={u}nait\.{e}, Danguol\.{e} and Zhang, Allen W. and Li, Jun and Lim, Jamie L. P. and Wu, Michele and Choi, Seongmin and Havasov, Eliyahu and Grewal, Diljot and Shi, Hongyu and Kim, Minsoo and Schwarz, Roland F. and Kaufmann, Tom and Dinh, Khanh N. and Uhlitz, Florian and Tran, Julie and Wu, Yushi and Patel, Ruchi and Ramakrishnan, Satish and Kim, DooA and Clarke, Justin and Green, Hunter and Ali, Emily and DiBona, Melody and Varice, Nancy and Kundra, Ritika and Broach, Vance and Gardner, Ginger J. and Roche, Kara L. and Sonoda, Yukio and Zivanovic, Oliver and Kim, Sarah H. and Grisham, Rachel N. and Liu, Ying L. and Viale, Agnes and Rusk, Nicole and Lakhman, Yulia and Ellenson, Lora H. and Tavar{\'e}, Simon and Aparicio, Samuel and Chi, Dennis S. and Aghajanian, Carol and Abu-Rustum, Nadeem R. and Friedman, Claire F. and Zamarin, Dmitriy and Weigelt, Britta and Bakhoum, Samuel F. and Shah, Sohrab P.}, journal = {Nature}, year = {2025}, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s41586-025-09240-3}, } -
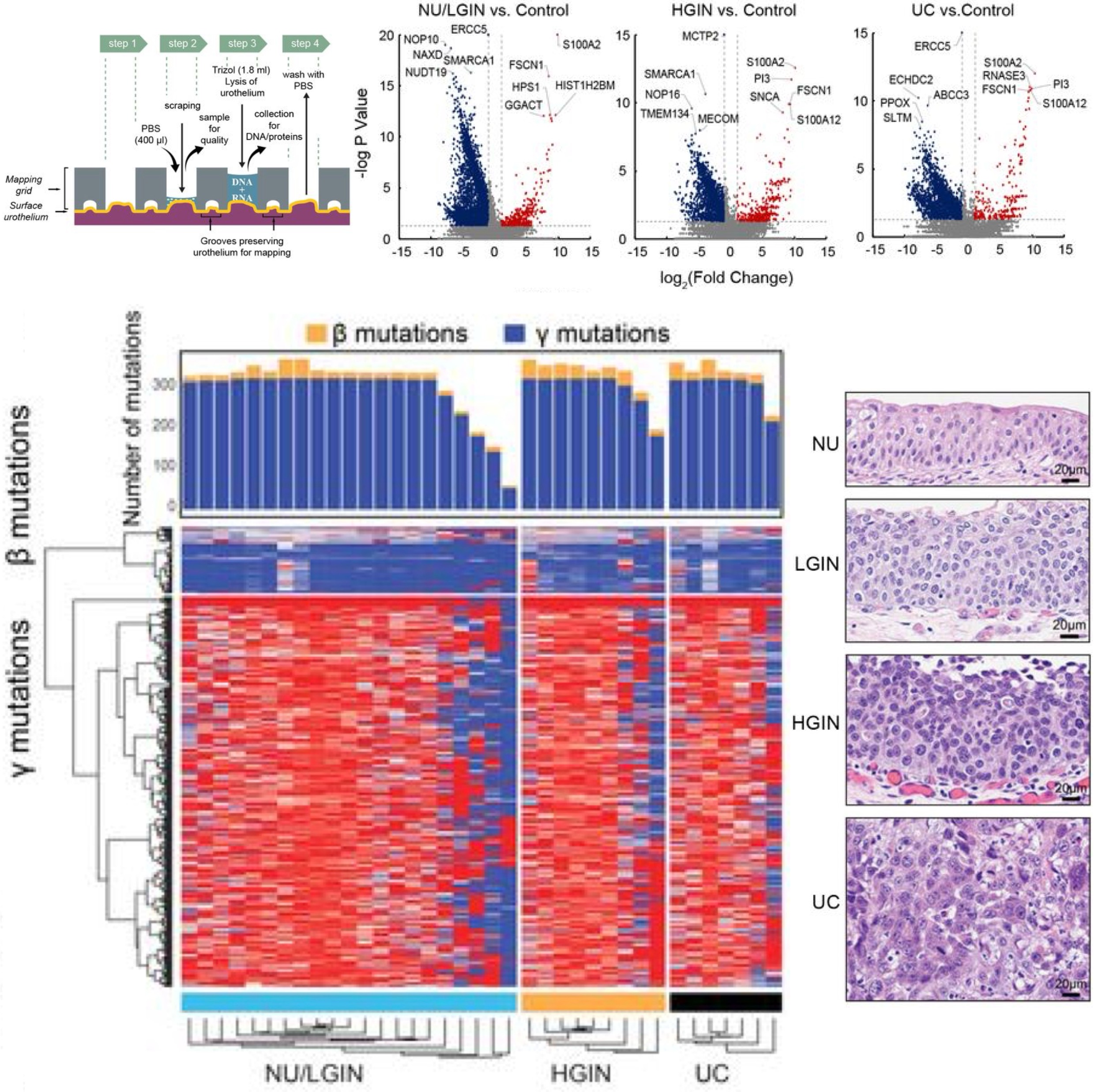 Dysregulated mitochondrial energy metabolism drives the progression of mucosal field effects to invasive bladder cancerSangkyou Lee, Sung Yun Jung, Pawel Kuś, Jolanta Bondaruk, June Goo Lee, Roman Jaksik, Nagireddy Putluri, Khanh N. Dinh, David Cogdell , Huiqin Chen, Yishan Wang , Jiansong Chen, Neema Navai, Colin Dinney, Cathy Mendelsohn, David McConkey, Richard R. Behringer, Charles C. Guo, Peng Wei, Marek Kimmel, and Bogdan CzerniakThe Journal of Pathology. Highlighted in Nature Reviews Urology , 2025
Dysregulated mitochondrial energy metabolism drives the progression of mucosal field effects to invasive bladder cancerSangkyou Lee, Sung Yun Jung, Pawel Kuś, Jolanta Bondaruk, June Goo Lee, Roman Jaksik, Nagireddy Putluri, Khanh N. Dinh, David Cogdell , Huiqin Chen, Yishan Wang , Jiansong Chen, Neema Navai, Colin Dinney, Cathy Mendelsohn, David McConkey, Richard R. Behringer, Charles C. Guo, Peng Wei, Marek Kimmel, and Bogdan CzerniakThe Journal of Pathology. Highlighted in Nature Reviews Urology , 2025Multiplatform mutational and gene expression profiling complemented with proteomic and metabolomic spatial mapping were used on the whole-organ scale to identify the molecular profile of bladder cancer evolution from field effects. Analysis of the mutational landscape identified three types of mutations, referred to as alpha, beta, and gamma. Time modeling of the mutations revealed that carcinogenesis may span 30 years and can be divided into dormant and progressive phases. The alpha mutations developed in the dormant phase. The progressive phase lasted 5 years and was signified by expanding beta mutations, but it was driven to invasive cancer by gamma mutations. The mutational landscape emerged on a background of disorganized urothelial differentiation, activated epithelial-mesenchymal transition, and enhanced immune infiltration with T-cell exhaustion. Complex dysregulation of mitochondrial energy metabolism with downregulation of oxidative phosphorylation emerged as the leading mechanism driving the progression of mucosal field effects to invasive cancer.
@article{lee2025dysregulated, dimensions = {true}, title = {Dysregulated mitochondrial energy metabolism drives the progression of mucosal field effects to invasive bladder cancer}, author = {Lee, Sangkyou and Jung, Sung Yun and Ku{\'s}, Pawel and Bondaruk, Jolanta and Lee, June Goo and Jaksik, Roman and Putluri, Nagireddy and Dinh, Khanh N. and Cogdell, David and Chen, Huiqin and Wang, Yishan and Chen, Jiansong and Navai, Neema and Dinney, Colin and Mendelsohn, Cathy and McConkey, David and Behringer, Richard R. and Guo, Charles C. and Wei, Peng and Kimmel, Marek and Czerniak, Bogdan}, journal = {The Journal of Pathology}, year = {2025}, publisher = {Pathological Society}, doi = {10.1002/path.6474}, } -
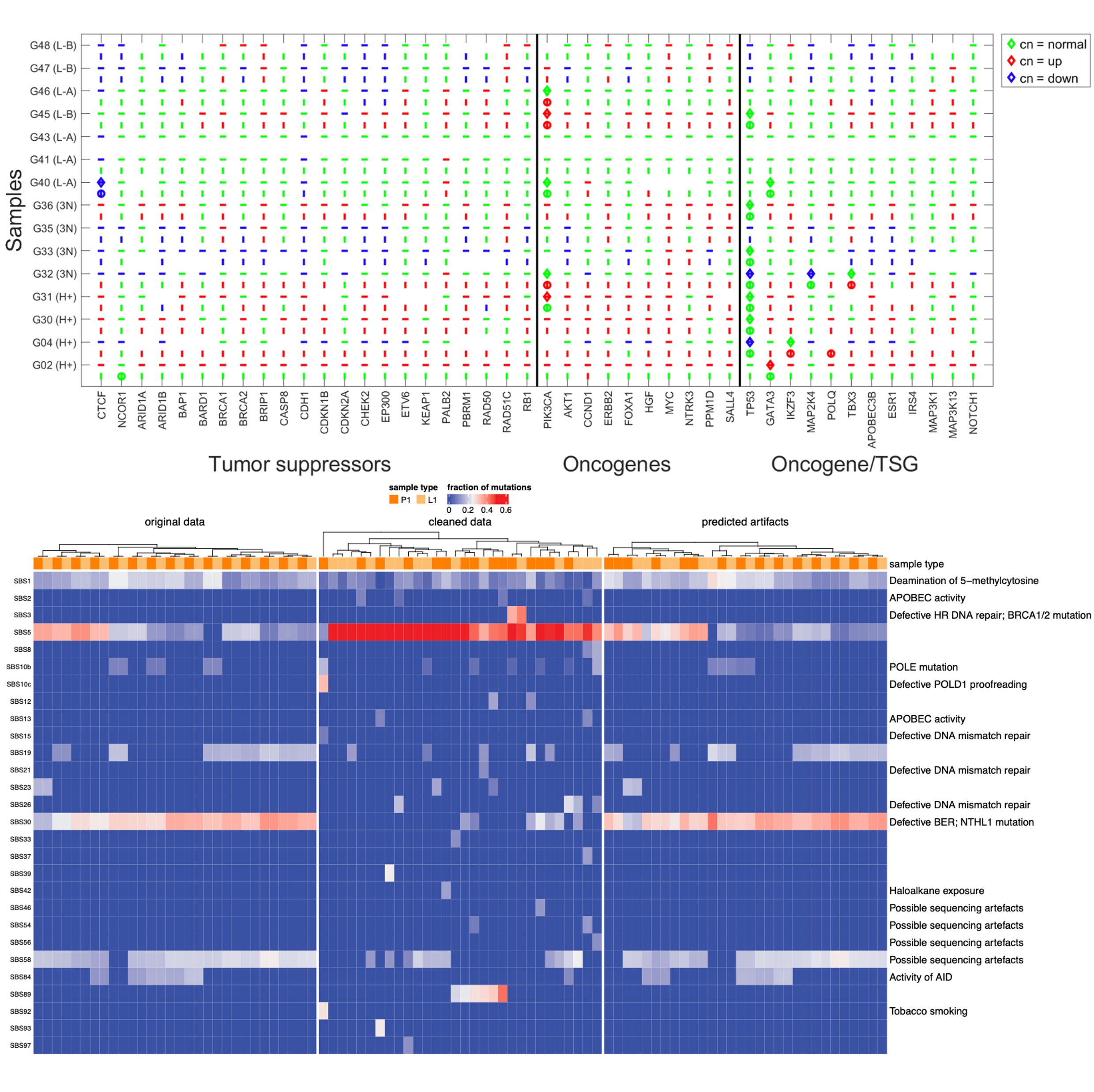 Statistical and evolutionary analysis of sequenced DNA from breast cancer FFPE specimensMonika K. Kurpas, Pawel Kuś, Roman Jaksik, Khanh N. Dinh, Agnieszka Adamczyk, Kaja Majchrzyk, and Marek KimmelbioRxiv, 2025
Statistical and evolutionary analysis of sequenced DNA from breast cancer FFPE specimensMonika K. Kurpas, Pawel Kuś, Roman Jaksik, Khanh N. Dinh, Agnieszka Adamczyk, Kaja Majchrzyk, and Marek KimmelbioRxiv, 2025Despite the introduction of instant freezing of tumor specimens, formalin-fixed paraffin-embedded (FFPE) blocks of tissue are still commonplace in clinical practice and constitute an important reference for genetic epidemiology of cancer. We carried out a study of a collection of breast tumors paired with lymph-node metastases and analyzed using advanced computational methods, to determine how much information can be obtained from mid-depth whole-exome bulk DNA sequencing. We gathered 15 paired (primary and an involved lymph node) excised breast tumors of different molecular subtypes (HER2+, triple negative, luminal A and luminal B HER2-), from the National Research Institute of Oncology, Krakow (Poland) Branch. FFPE specimens contained typical artifacts, manifesting themselves in spurious DNA variant calls. We used several bioinformatics tools to remove the artifacts and analyzed the exomic data, using both commercial and original in-house computational techniques. We used several of recent bioinformatics tools to remove the FFPE artifacts and found a serious dispersal of outcomes. After calibration, a series of analyses was performed, including copy number study, resulting in ploidy levels ranging from 1 to 5 (average of 2.5). Positive association was found between the frequency of oncogenes relative to tumor suppressor genes and DNA copy number. In addition, we carried out analyses of the clonal structure of the data using original computational methods based on evolutionary modeling. Interesting results concerning clonal structure, early tumor expansion, and interdependence of the primary tumor and lymph node metastases have been obtained. Despite the imperfections of the FFPE data, many important features of molecular evolution of tumor DNA can be recovered from routine clinical samples.
@article{kurpas2025statistical, dimensions = {true}, title = {Statistical and evolutionary analysis of sequenced DNA from breast cancer FFPE specimens}, author = {Kurpas, Monika K. and Ku{\'s}, Pawel and Jaksik, Roman and Dinh, Khanh N. and Adamczyk, Agnieszka and Majchrzyk, Kaja and Kimmel, Marek}, journal = {bioRxiv}, year = {2025}, doi = {10.1101/2025.10.04.680485}, } -
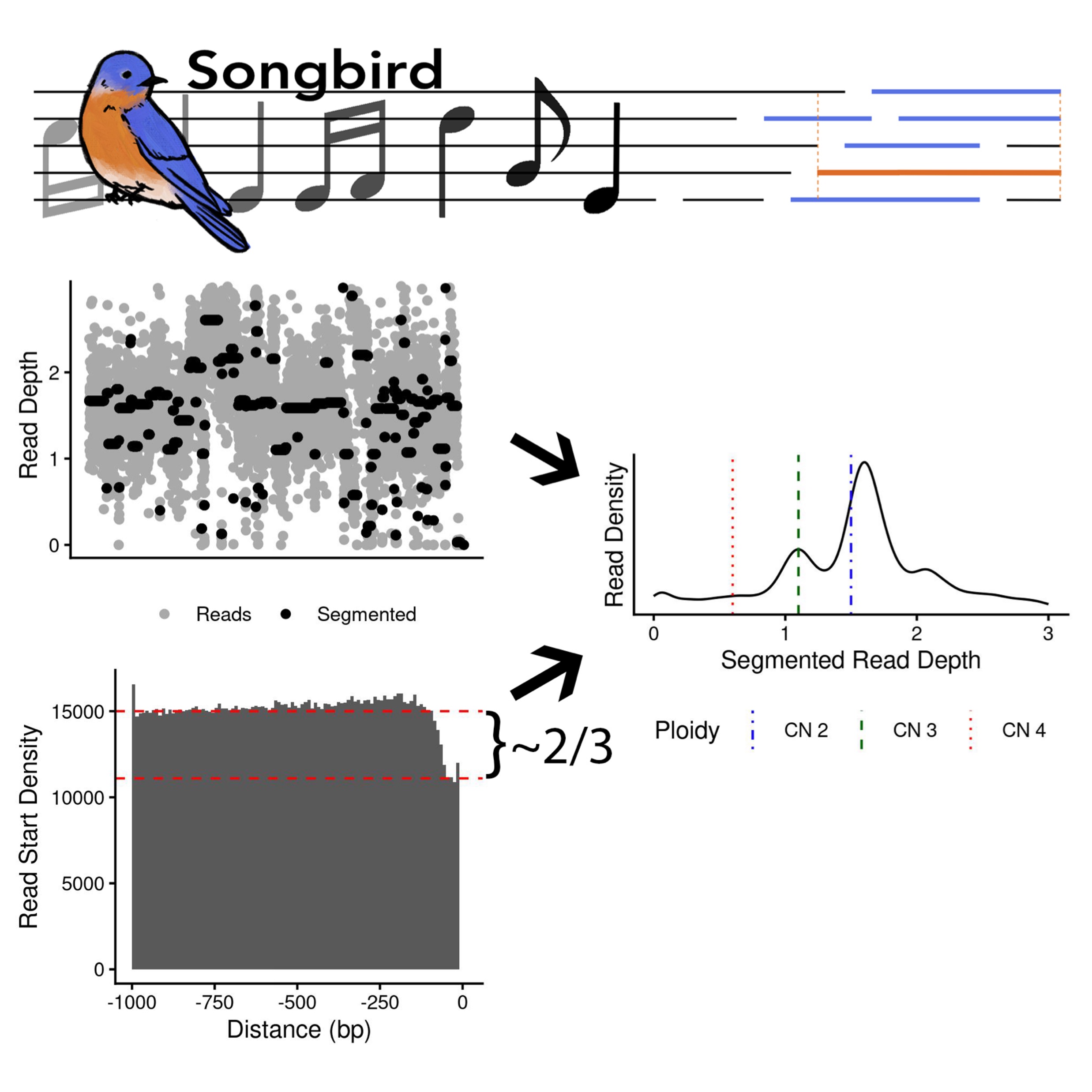 Wavelet based whole genome doubling aware single cell copy number callingBenjamin K. Wesley, Frank Wos, Soren Germer, Jade E. B. Carter, Silas Maniatis, Khanh N. Dinh, Timothy Chu, Nicolas Robine, Rebecca Fitzgerald, Lizhe Zhuang, Simon Tavaré, and Karol Nowicki-OsuchbioRxiv, 2025
Wavelet based whole genome doubling aware single cell copy number callingBenjamin K. Wesley, Frank Wos, Soren Germer, Jade E. B. Carter, Silas Maniatis, Khanh N. Dinh, Timothy Chu, Nicolas Robine, Rebecca Fitzgerald, Lizhe Zhuang, Simon Tavaré, and Karol Nowicki-OsuchbioRxiv, 2025Advances in single cell whole genome sequencing enable profiling of the copy number state of thousands of cells with little sequencing bias across the genome. The Direct Library Preparation + (DLP+) technique is a whole genome amplification-free single cell whole genome sequencing method that achieves high throughput by fragmenting each cell’s genome and ligating sequencing adapters using a modified Tn5 transposase, and sequencing to low coverage (less than 0.1x). Despite recent advances in experimental approaches, data analysis of scDNA lags behind and the existing methods are not optimized for the analysis of frozen samples with variable DNA preservation. Furthermore, existing tools predominantly rely on read depth ratio in predefined genomic bins to call copy number, making whole genome detection unidentifiable. To address this, we introduce Songbird, a single cell whole genome sequencing copy number caller that is whole genome duplication sensitive, and outperforms existing tools both in breakpoint identification and true copy number detection. We demonstrate that Songbird is robust down to extremely low coverage (less than 0.01x), adaptable to a variety of genome versions (hg19, hg38, hs.1), and is extensible to other single cell whole genome sequencing methods that rely on Tn5 tagmentation to fragment the genome.
@article{wesley2025wavelet, dimensions = {true}, title = {Wavelet based whole genome doubling aware single cell copy number calling}, author = {Wesley, Benjamin K. and Wos, Frank and Germer, Soren and Carter, Jade E. B. and Maniatis, Silas and Dinh, Khanh N. and Chu, Timothy and Robine, Nicolas and Fitzgerald, Rebecca and Zhuang, Lizhe and Tavar{\'e}, Simon and Nowicki-Osuch, Karol}, journal = {bioRxiv}, year = {2025}, doi = {10.64898/2025.12.18.693686}, }
2024
-
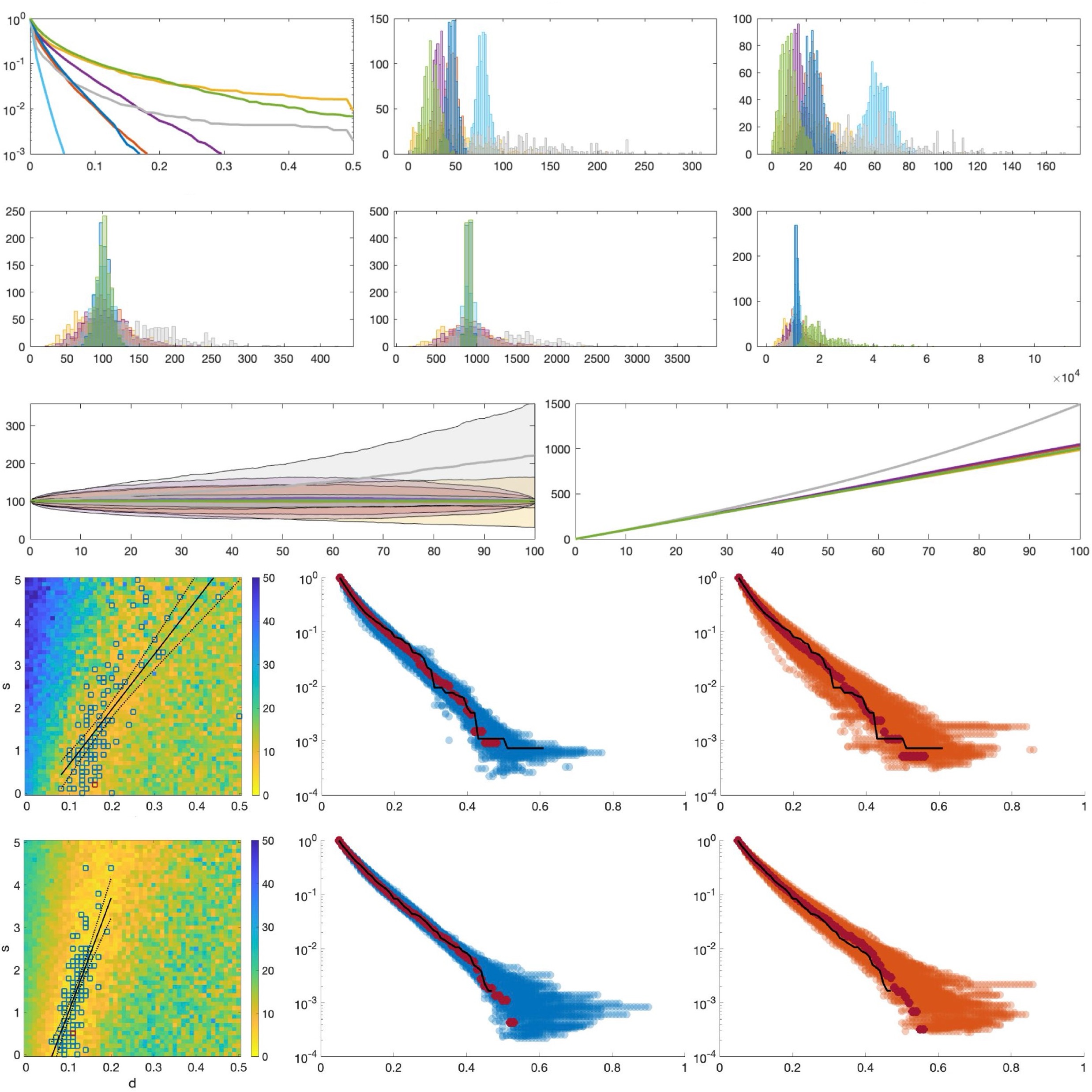 Comparison of tug-of-war models assuming Moran versus branching process population dynamicsKhanh N. Dinh, Monika K. Kurpas, and Marek KimmeleLife, 2024
Comparison of tug-of-war models assuming Moran versus branching process population dynamicsKhanh N. Dinh, Monika K. Kurpas, and Marek KimmeleLife, 2024Mutations arising during cancer evolution are typically categorized as either ’drivers’ or ’passengers’, depending on whether they increase the cell fitness. Recently, McFarland et al. introduced the Tug-of-War model for the joint effect of rare advantageous drivers and frequent but deleterious passengers. We examine this model under two common but distinct frameworks, the Moran model and the branching process. We show that frequently used statistics are similar between a version of the Moran model and the branching process conditioned on the final cell count, under different selection scenarios. We infer the selection coefficients for three breast cancer samples, resulting in good fits of the shape of their Site Frequency Spectra. All fitted values for the selective disadvantage of passenger mutations are nonzero, supporting the view that they exert deleterious selection during tumorigenesis that driver mutations must compensate.
@article{dinh2024comparison, dimensions = {true}, title = {Comparison of tug-of-war models assuming Moran versus branching process population dynamics}, author = {Dinh, Khanh N. and Kurpas, Monika K. and Kimmel, Marek}, journal = {eLife}, volume = {13}, year = {2024}, publisher = {eLife Sciences Publications Limited}, doi = {10.7554/eLife.94597.1}, } -
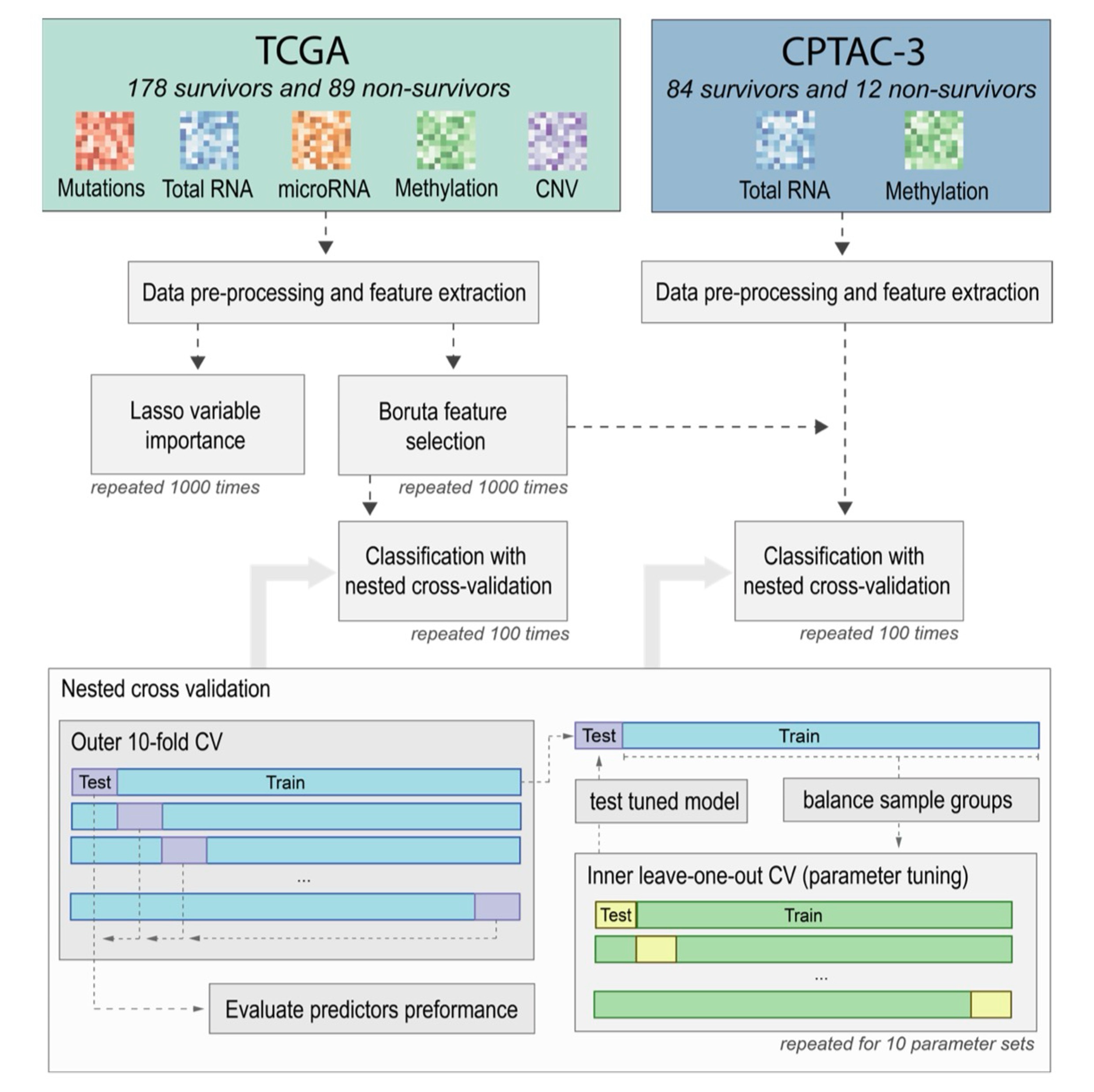 Multiomics-based feature extraction and selection for the prediction of lung cancer survivalRoman Jaksik, Kamila Szumała, Khanh N. Dinh, and Jarosław ŚmiejaInternational Journal of Molecular Sciences, 2024
Multiomics-based feature extraction and selection for the prediction of lung cancer survivalRoman Jaksik, Kamila Szumała, Khanh N. Dinh, and Jarosław ŚmiejaInternational Journal of Molecular Sciences, 2024Lung cancer is a global health challenge, hindered by delayed diagnosis and the disease’s complex molecular landscape. Accurate patient survival prediction is critical, motivating the exploration of various -omics datasets using machine learning methods. Leveraging multi-omics data, this study seeks to enhance the accuracy of survival prediction by proposing new feature extraction techniques combined with unbiased feature selection. Two lung adenocarcinoma multi-omics datasets, originating from the TCGA and CPTAC-3 projects, were employed for this purpose, emphasizing gene expression, methylation, and mutations as the most relevant data sources that provide features for the survival prediction models. Additionally, gene set aggregation was shown to be the most effective feature extraction method for mutation and copy number variation data. Using the TCGA dataset, we identified 32 molecular features that allowed the construction of a 2-year survival prediction model with an AUC of 0.839. The selected features were additionally tested on an independent CPTAC-3 dataset, achieving an AUC of 0.815 in nested cross-validation, which confirmed the robustness of the identified features.
@article{jaksik2024multiomics, dimensions = {true}, title = {Multiomics-based feature extraction and selection for the prediction of lung cancer survival}, author = {Jaksik, Roman and Szuma{\l}a, Kamila and Dinh, Khanh N. and {\'S}mieja, Jaros{\l}aw}, journal = {International Journal of Molecular Sciences}, volume = {25}, number = {7}, pages = {3661}, year = {2024}, publisher = {MDPI}, doi = {10.3390/ijms25073661}, } -
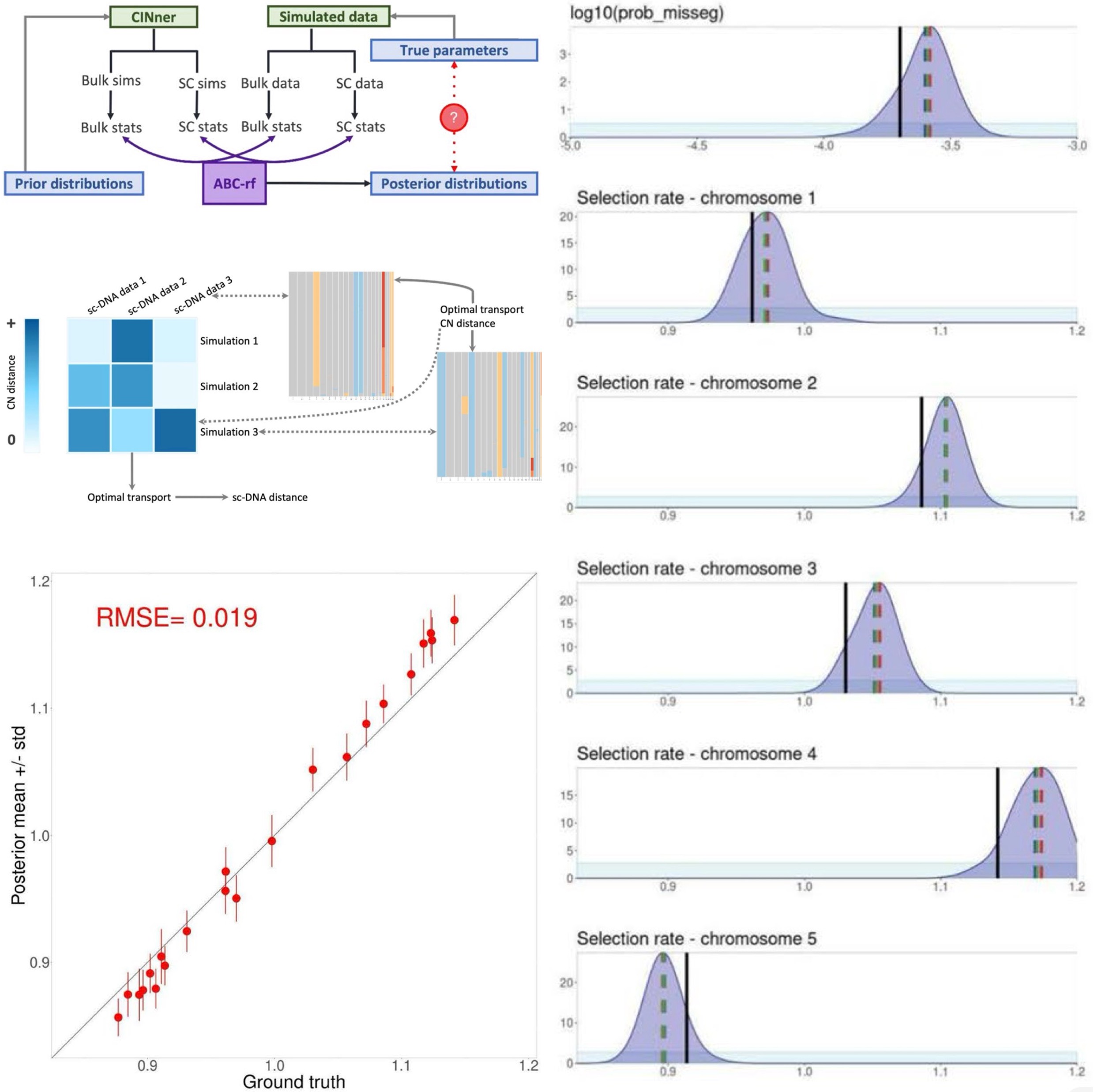 Inference of chromosome selection parameters and missegregation rate in cancer from DNA-sequencing dataZijin Xiang , Zhihan Liu, and Khanh N. DinhScientific Reports, 2024
Inference of chromosome selection parameters and missegregation rate in cancer from DNA-sequencing dataZijin Xiang , Zhihan Liu, and Khanh N. DinhScientific Reports, 2024Aneuploidy is frequently observed in cancers and has been linked to poor patient outcome. Analysis of aneuploidy in DNA-sequencing (DNA-seq) data necessitates untangling the effects of the Copy Number Aberration (CNA) occurrence rates and the selection coefficients that act upon the resulting karyotypes. We introduce a parameter inference algorithm that takes advantage of both bulk and single-cell DNA-seq cohorts. The method is based on Approximate Bayesian Computation (ABC) and utilizes CINner, our recently introduced simulation algorithm of chromosomal instability in cancer. We examine three groups of statistics to summarize the data in the ABC routine: (A) Copy Number-based measures, (B) phylogeny tip statistics, and (C) phylogeny balance indices. Using these statistics, our method can recover both the CNA probabilities and selection parameters from ground truth data, and performs well even for data cohorts of relatively small sizes. We find that only statistics in groups A and C are well-suited for identifying CNA probabilities, and only group A carries the signals for estimating selection parameters. Moreover, the low number of CNA events at large scale compared to cell counts in single-cell samples means that statistics in group B cannot be estimated accurately using phylogeny reconstruction algorithms at the chromosome level. As data from both bulk and single-cell DNA-sequencing techniques becomes increasingly available, our inference framework promises to facilitate the analysis of distinct cancer types, differentiation between selection and neutral drift, and prediction of cancer clonal dynamics.
@article{xiang2024inference, dimensions = {true}, title = {Inference of chromosome selection parameters and missegregation rate in cancer from DNA-sequencing data}, author = {Xiang, Zijin and Liu, Zhihan and Dinh, Khanh N.}, journal = {Scientific Reports}, volume = {14}, number = {1}, pages = {17699}, year = {2024}, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s41598}, }
2023
-
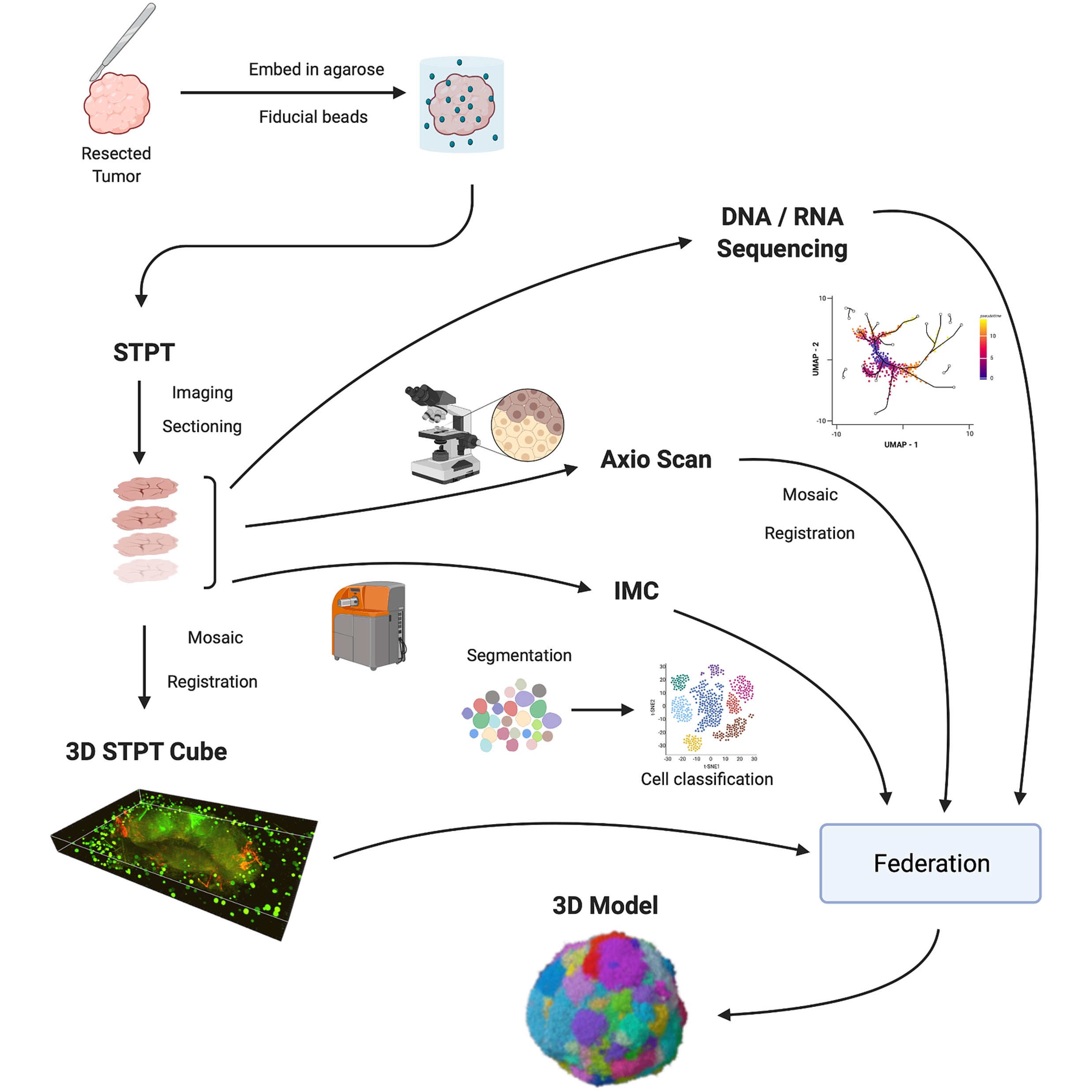 Imaging and Molecular Annotation of Xenographs and Tumours (IMAXT): high throughput data and analysis infrastructureEduardo A. González-Solares, Ali Dariush, Carlos González-Fernández, Aybüke Küpcü Yoldaş, Alireza Molaeinezhad, Mohammad Al Sa’d, Leigh Smith, Tristan Whitmarsh, Neil Millar, Nicholas Chornay, Ilaria Falciatori, Atefeh Fatemi, Daniel Goodwin, Laura Kuett, Claire M. Mulvey, Marta P. Ribes, Fatime Qosaj, Andrew Roth, Ignacio Vázquez-García, Spencer S. Watson, Jonas Windhager, Samuel Aparicio, Bernd Bodenmiller, Ed Boyden, Carlos Caldas, Owen Harris, Sohrab P. Shah, Simon Tavaré, Dario Bressan, Gregory J. Hannon, Nicholas A. Walton, and IMAXT ConsortiumBiological Imaging, 2023
Imaging and Molecular Annotation of Xenographs and Tumours (IMAXT): high throughput data and analysis infrastructureEduardo A. González-Solares, Ali Dariush, Carlos González-Fernández, Aybüke Küpcü Yoldaş, Alireza Molaeinezhad, Mohammad Al Sa’d, Leigh Smith, Tristan Whitmarsh, Neil Millar, Nicholas Chornay, Ilaria Falciatori, Atefeh Fatemi, Daniel Goodwin, Laura Kuett, Claire M. Mulvey, Marta P. Ribes, Fatime Qosaj, Andrew Roth, Ignacio Vázquez-García, Spencer S. Watson, Jonas Windhager, Samuel Aparicio, Bernd Bodenmiller, Ed Boyden, Carlos Caldas, Owen Harris, Sohrab P. Shah, Simon Tavaré, Dario Bressan, Gregory J. Hannon, Nicholas A. Walton, and IMAXT ConsortiumBiological Imaging, 2023With the aim of producing a 3D representation of tumors, imaging and molecular annotation of xenografts and tumors (IMAXT) uses a large variety of modalities in order to acquire tumor samples and produce a map of every cell in the tumor and its host environment. With the large volume and variety of data produced in the project, we developed automatic data workflows and analysis pipelines. We introduce a research methodology where scientists connect to a cloud environment to perform analysis close to where data are located, instead of bringing data to their local computers. Here, we present the data and analysis infrastructure, discuss the unique computational challenges and describe the analysis chains developed and deployed to generate molecularly annotated tumor models. Registration is achieved by use of a novel technique involving spherical fiducial marks that are visible in all imaging modalities used within IMAXT. The automatic pipelines are highly optimized and allow to obtain processed datasets several times quicker than current solutions narrowing the gap between data acquisition and scientific exploitation.
@article{gonzalez2023imaging, dimensions = {true}, title = {Imaging and Molecular Annotation of Xenographs and Tumours (IMAXT): high throughput data and analysis infrastructure}, author = {Gonz{\'a}lez-Solares, Eduardo A. and Dariush, Ali and Gonz{\'a}lez-Fern{\'a}ndez, Carlos and Yolda{\c{s}}, Ayb{\"u}ke K{\"u}pc{\"u} and Molaeinezhad, Alireza and Al Sa'd, Mohammad and Smith, Leigh and Whitmarsh, Tristan and Millar, Neil and Chornay, Nicholas and Falciatori, Ilaria and Fatemi, Atefeh and Goodwin, Daniel and Kuett, Laura and Mulvey, Claire M. and Ribes, Marta P. and Qosaj, Fatime and Roth, Andrew and Vázquez-García, Ignacio and Watson, Spencer S. and Windhager, Jonas and Aparicio, Samuel and Bodenmiller, Bernd and Boyden, Ed and Caldas, Carlos and Harris, Owen and Shah, Sohrab P. and Tavar{\'e}, Simon and Bressan, Dario and Hannon, Gregory J. and Walton, Nicholas A. and Consortium, IMAXT}, journal = {Biological Imaging}, volume = {3}, pages = {e11}, year = {2023}, publisher = {Cambridge University Press}, doi = {10.1017/S2633903X23000090}, }
2022
-
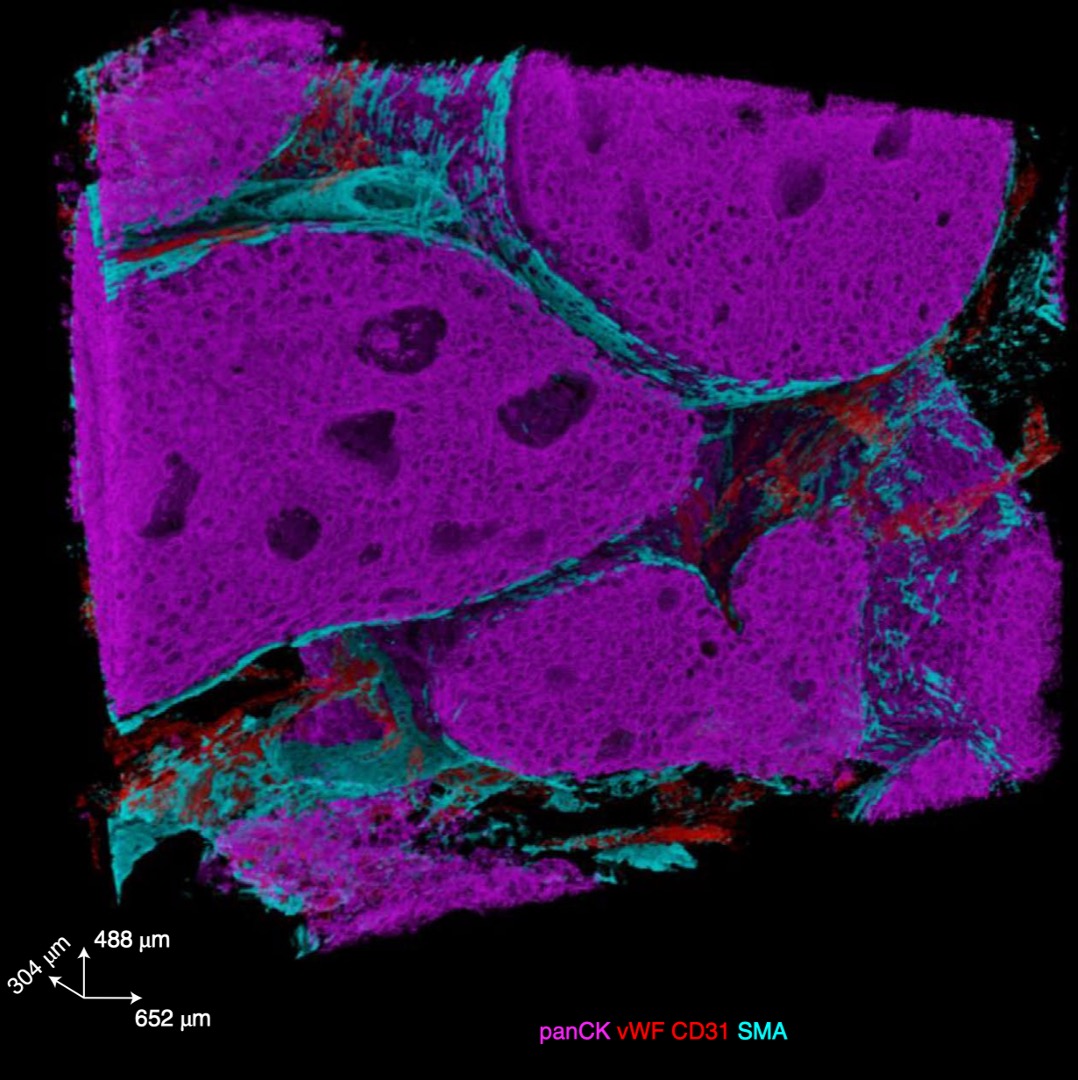 Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironmentLaura Kuett, Raúl Catena, Alaz Özcan, Alex Plüss, IMAXT Consortium, Peter Schraml, Holger Moch, Natalie Souza, and Bernd BodenmillerNature Cancer, 2022
Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironmentLaura Kuett, Raúl Catena, Alaz Özcan, Alex Plüss, IMAXT Consortium, Peter Schraml, Holger Moch, Natalie Souza, and Bernd BodenmillerNature Cancer, 2022A holistic understanding of tissue and organ structure and function requires the detection of molecular constituents in their original three-dimensional (3D) context. Imaging mass cytometry (IMC) enables simultaneous detection of up to 40 antigens and transcripts using metal-tagged antibodies but has so far been restricted to two-dimensional imaging. Here we report the development of 3D IMC for multiplexed 3D tissue analysis at single-cell resolution and demonstrate the utility of the technology by analysis of human breast cancer samples. The resulting 3D models reveal cellular and microenvironmental heterogeneity and cell-level tissue organization not detectable in two dimensions. 3D IMC will prove powerful in the study of phenomena occurring in 3D space such as tumor cell invasion and is expected to provide invaluable insights into cellular microenvironments and tissue architecture.
@article{kuett2022three, dimensions = {true}, title = {Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironment}, author = {Kuett, Laura and Catena, Ra{\'u}l and {\"O}zcan, Alaz and Pl{\"u}ss, Alex and Consortium, IMAXT and Schraml, Peter and Moch, Holger and de Souza, Natalie and Bodenmiller, Bernd}, journal = {Nature Cancer}, volume = {3}, number = {1}, pages = {122--133}, year = {2022}, publisher = {Nature Publishing Group US New York}, doi = {10.1038/s43018-021-00301-w}, } -
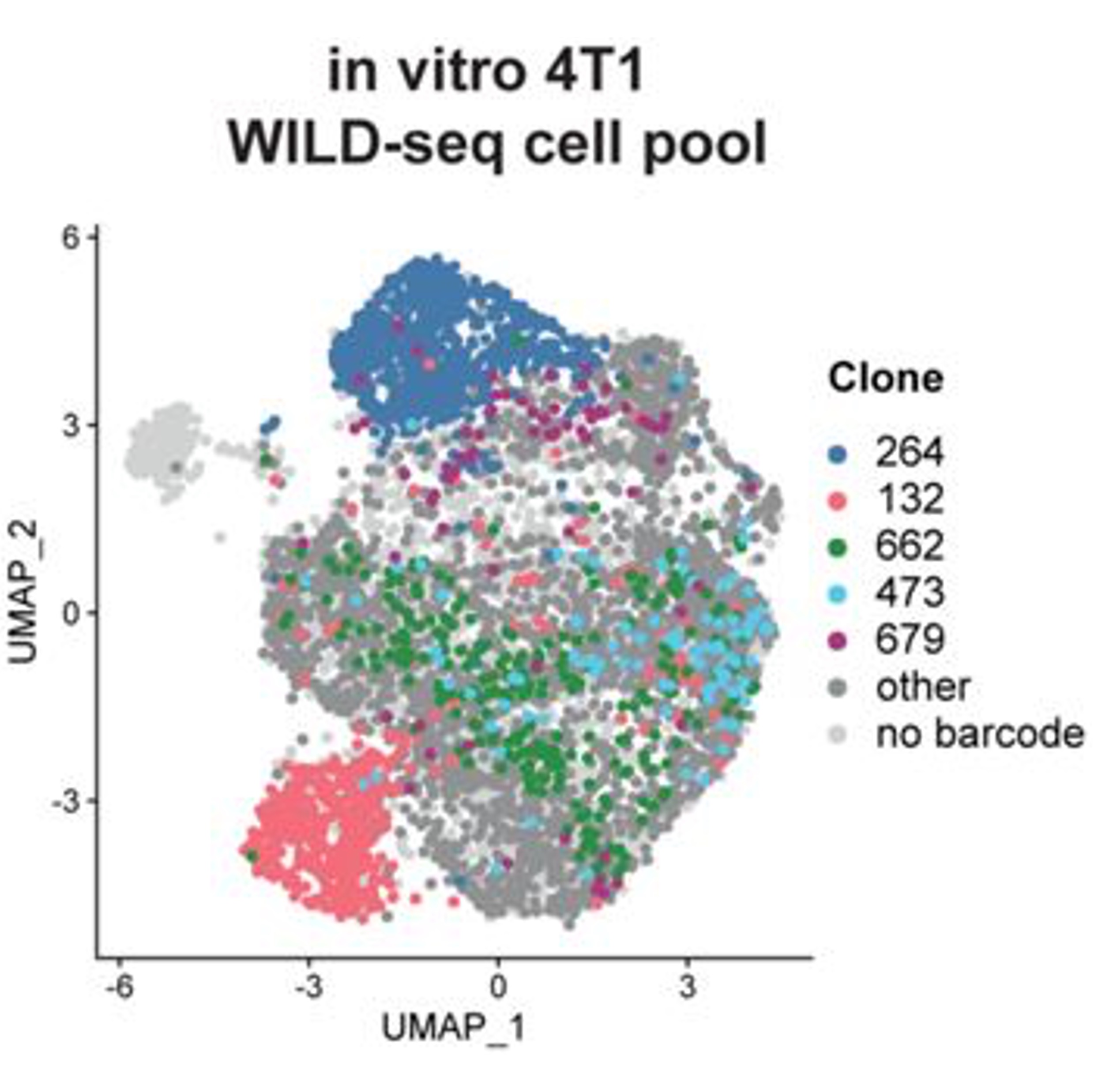 Clonal transcriptomics identifies mechanisms of chemoresistance and empowers rational design of combination therapiesSophia A. Wild, Ian G. Cannell, Ashley Nicholls, Katarzyna Kania, Dario Bressan, IMAXT Consortium, Gregory J. Hannon, and Kirsty SawickaeLife, 2022
Clonal transcriptomics identifies mechanisms of chemoresistance and empowers rational design of combination therapiesSophia A. Wild, Ian G. Cannell, Ashley Nicholls, Katarzyna Kania, Dario Bressan, IMAXT Consortium, Gregory J. Hannon, and Kirsty SawickaeLife, 2022Tumour heterogeneity is thought to be a major barrier to successful cancer treatment due to the presence of drug resistant clonal lineages. However, identifying the characteristics of such lineages that underpin resistance to therapy has remained challenging. Here, we utilise clonal transcriptomics with WILD-seq; Wholistic Interrogation of Lineage Dynamics by sequencing, in mouse models of triple-negative breast cancer (TNBC) to understand response and resistance to therapy, including BET bromodomain inhibition and taxane-based chemotherapy. These analyses revealed oxidative stress protection by NRF2 as a major mechanism of taxane resistance and led to the discovery that our tumour models are collaterally sensitive to asparagine deprivation therapy using the clinical stage drug L-asparaginase after frontline treatment with docetaxel. In summary, clonal transcriptomics with WILD-seq identifies mechanisms of resistance to chemotherapy that are also operative in patients and pin points asparagine bioavailability as a druggable vulnerability of taxane-resistant lineages.
@article{wild2022clonal, dimensions = {true}, title = {Clonal transcriptomics identifies mechanisms of chemoresistance and empowers rational design of combination therapies}, author = {Wild, Sophia A. and Cannell, Ian G. and Nicholls, Ashley and Kania, Katarzyna and Bressan, Dario and Consortium, IMAXT and Hannon, Gregory J. and Sawicka, Kirsty}, journal = {eLife}, volume = {11}, pages = {e80981}, year = {2022}, publisher = {eLife Sciences Publications, Ltd}, doi = {10.7554/eLife.80981}, } -
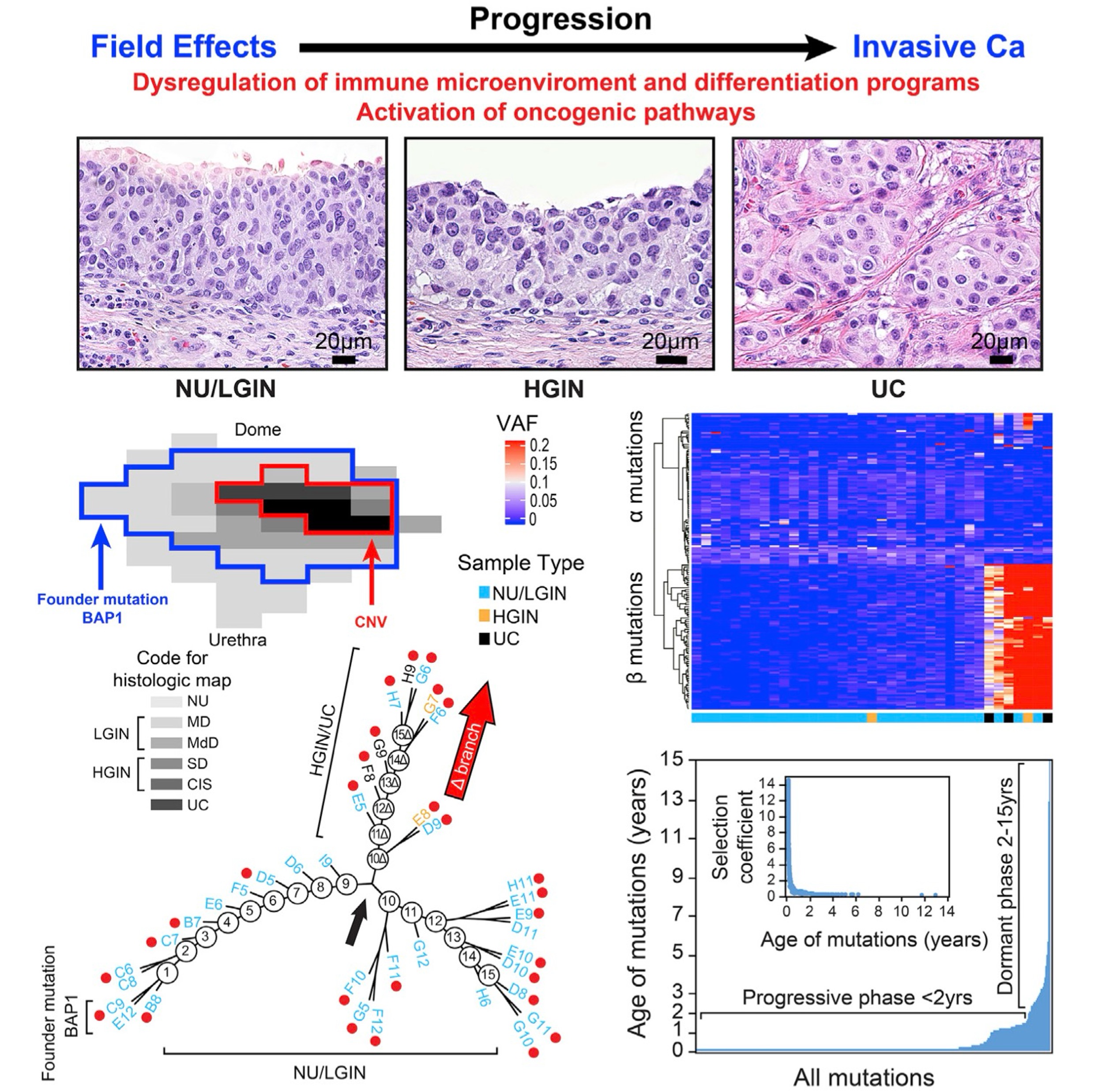 The origin of bladder cancer from mucosal field effectsJolanta Bondaruk, Roman Jaksik, Ziqiao Wang, David Cogdell, Sangkyou Lee , Yujie Chen, Khanh N. Dinh, Tadeusz Majewski, Li Zhang, Shaolong Cao, Feng Tian, Hui Yao, Pawel Kuś , Huiqin Chen, John N. Weinstein, Neema Navai, Colin Dinney, Jianjun Gao, Dan Theodorescu, Christopher Logothetis, Charles C. Guo, Wenyi Wang, David McConkey, Peng Wei, Marek Kimmel, and Bogdan CzerniakiScience, 2022
The origin of bladder cancer from mucosal field effectsJolanta Bondaruk, Roman Jaksik, Ziqiao Wang, David Cogdell, Sangkyou Lee , Yujie Chen, Khanh N. Dinh, Tadeusz Majewski, Li Zhang, Shaolong Cao, Feng Tian, Hui Yao, Pawel Kuś , Huiqin Chen, John N. Weinstein, Neema Navai, Colin Dinney, Jianjun Gao, Dan Theodorescu, Christopher Logothetis, Charles C. Guo, Wenyi Wang, David McConkey, Peng Wei, Marek Kimmel, and Bogdan CzerniakiScience, 2022Whole-organ mapping was used to study molecular changes in the evolution of bladder cancer from field effects. We identified more than 100 dysregulated pathways, involving immunity, differentiation, and transformation, as initiators of carcinogenesis. Dysregulation of interleukins signified the involvement of inflammation in the incipient phases of the process. An aberrant methylation/expression of multiple HOX genes signified dysregulation of the differentiation program. We identified three types of mutations based on their geographic distribution. The most common were mutations restricted to individual mucosal samples that targeted uroprogenitor cells. Two types of mutations were associated with clonal expansion and involved large areas of mucosa. The αmutations occurred at low frequencies while the βmutations increased in frequency with disease progression. Modeling revealed that bladder carcinogenesis spans 10-15 years and can be divided into dormant and progressive phases. The progressive phase lasted 1-2 years and was driven by βmutations.
@article{bondaruk2022origin, dimensions = {true}, title = {The origin of bladder cancer from mucosal field effects}, author = {Bondaruk, Jolanta and Jaksik, Roman and Wang, Ziqiao and Cogdell, David and Lee, Sangkyou and Chen, Yujie and Dinh, Khanh N. and Majewski, Tadeusz and Zhang, Li and Cao, Shaolong and Tian, Feng and Yao, Hui and Ku{\'s}, Pawel and Chen, Huiqin and Weinstein, John N. and Navai, Neema and Dinney, Colin and Gao, Jianjun and Theodorescu, Dan and Logothetis, Christopher and Guo, Charles C. and Wang, Wenyi and McConkey, David and Wei, Peng and Kimmel, Marek and Czerniak, Bogdan}, journal = {iScience}, volume = {25}, number = {7}, year = {2022}, publisher = {Elsevier}, doi = {10.1016/j.isci.2022.104551}, } -
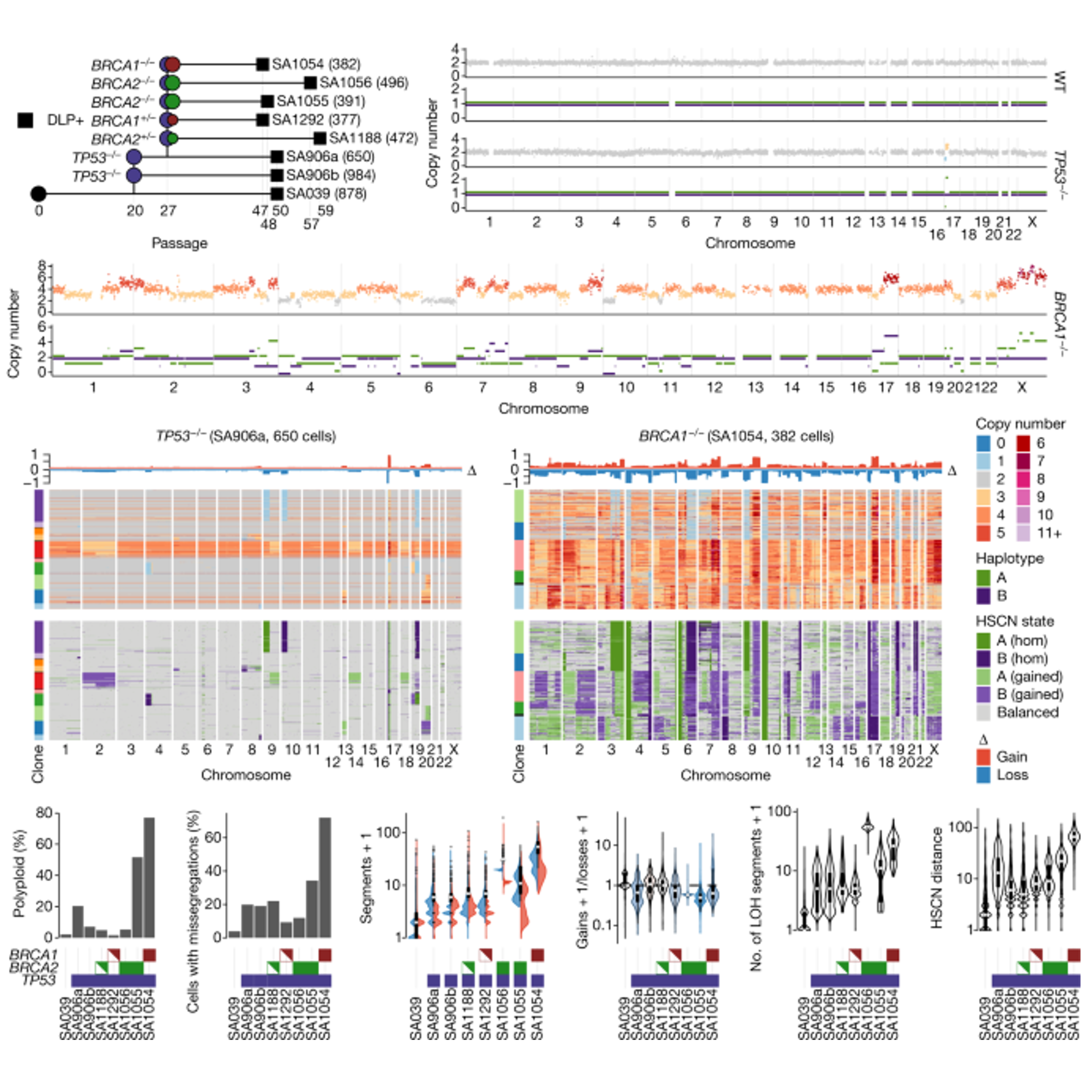 Single-cell genomic variation induced by mutational processes in cancerTyler Funnell, Ciara H O’Flanagan, Marc J Williams, Andrew McPherson, Steven McKinney, Farhia Kabeer, Hakwoo Lee, Sohrab Salehi, Ignacio Vázquez-García, Hongyu Shi, Emily Leventhal, Tehmina Masud, Peter Eirew, Damian Yap, Allen W. Zhang, Jamie L. P. Lim, Beixi Wang, Jazmine Brimhall, Justina Biele, Jerome Ting, Vinci Au, Michael Van Vliet , Yi Fei Liu, Sean Beatty, Daniel Lai, Jenifer Pham, Diljot Grewal, Douglas Abrams, Eliyahu Havasov, Samantha Leung, Viktoria Bojilova, Richard A. Moore, Nicole Rusk, Florian Uhlitz, Nicholas Ceglia, Adam C. Weiner, Elena Zaikova, J. Maxwell Douglas, Dmitriy Zamarin, Britta Weigelt, Sarah H. Kim, Arnaud Da Cruz Paula, Jorge S. Reis-Filho, Spencer D. Martin, Yangguang Li, Hong Xu, Teresa Ruiz Algara, So Ra Lee, Viviana Cerda Llanos, David G. Huntsman, Jessica N. McAlpine, IMAXT Consortium, Sohrab P. Shah, and Samuel AparicioNature, 2022
Single-cell genomic variation induced by mutational processes in cancerTyler Funnell, Ciara H O’Flanagan, Marc J Williams, Andrew McPherson, Steven McKinney, Farhia Kabeer, Hakwoo Lee, Sohrab Salehi, Ignacio Vázquez-García, Hongyu Shi, Emily Leventhal, Tehmina Masud, Peter Eirew, Damian Yap, Allen W. Zhang, Jamie L. P. Lim, Beixi Wang, Jazmine Brimhall, Justina Biele, Jerome Ting, Vinci Au, Michael Van Vliet , Yi Fei Liu, Sean Beatty, Daniel Lai, Jenifer Pham, Diljot Grewal, Douglas Abrams, Eliyahu Havasov, Samantha Leung, Viktoria Bojilova, Richard A. Moore, Nicole Rusk, Florian Uhlitz, Nicholas Ceglia, Adam C. Weiner, Elena Zaikova, J. Maxwell Douglas, Dmitriy Zamarin, Britta Weigelt, Sarah H. Kim, Arnaud Da Cruz Paula, Jorge S. Reis-Filho, Spencer D. Martin, Yangguang Li, Hong Xu, Teresa Ruiz Algara, So Ra Lee, Viviana Cerda Llanos, David G. Huntsman, Jessica N. McAlpine, IMAXT Consortium, Sohrab P. Shah, and Samuel AparicioNature, 2022How cell-to-cell copy number alterations that underpin genomic instability in human cancers drive genomic and phenotypic variation, and consequently the evolution of cancer, remains understudied. Here, by applying scaled single-cell whole-genome sequencing to wild-type, TP53-deficient and TP53-deficient; BRCA1-deficient or TP53-deficient; BRCA2-deficient mammary epithelial cells (13,818 genomes), and to primary triple-negative breast cancer (TNBC) and high-grade serous ovarian cancer (HGSC) cells (22,057 genomes), we identify three distinct ’foreground’ mutational patterns that are defined by cell-to-cell structural variation. Cell- and clone-specific high-level amplifications, parallel haplotype-specific copy number alterations and copy number segment length variation (serrate structural variations) had measurable phenotypic and evolutionary consequences. In TNBC and HGSC, clone-specific high-level amplifications in known oncogenes were highly prevalent in tumours bearing fold-back inversions, relative to tumours with homologous recombination deficiency, and were associated with increased clone-to-clone phenotypic variation. Parallel haplotype-specific alterations were also commonly observed, leading to phylogenetic evolutionary diversity and clone-specific mono-allelic expression. Serrate variants were increased in tumours with fold-back inversions and were highly correlated with increased genomic diversity of cellular populations. Together, our findings show that cell-to-cell structural variation contributes to the origins of phenotypic and evolutionary diversity in TNBC and HGSC, and provide insight into the genomic and mutational states of individual cancer cells.
@article{funnell2022single, dimensions = {true}, title = {Single-cell genomic variation induced by mutational processes in cancer}, author = {Funnell, Tyler and O'Flanagan, Ciara H and Williams, Marc J and McPherson, Andrew and McKinney, Steven and Kabeer, Farhia and Lee, Hakwoo and Salehi, Sohrab and Vázquez-García, Ignacio and Shi, Hongyu and Leventhal, Emily and Masud, Tehmina and Eirew, Peter and Yap, Damian and Zhang, Allen W. and Lim, Jamie L. P. and Wang, Beixi and Brimhall, Jazmine and Biele, Justina and Ting, Jerome and Au, Vinci and Van Vliet, Michael and Liu, Yi Fei and Beatty, Sean and Lai, Daniel and Pham, Jenifer and Grewal, Diljot and Abrams, Douglas and Havasov, Eliyahu and Leung, Samantha and Bojilova, Viktoria and Moore, Richard A. and Rusk, Nicole and Uhlitz, Florian and Ceglia, Nicholas and Weiner, Adam C. and Zaikova, Elena and Douglas, J. Maxwell and Zamarin, Dmitriy and Weigelt, Britta and Kim, Sarah H. and Da Cruz Paula, Arnaud and Reis-Filho, Jorge S. and Martin, Spencer D. and Li, Yangguang and Xu, Hong and de Algara, Teresa Ruiz and Lee, So Ra and Llanos, Viviana Cerda and Huntsman, David G. and McAlpine, Jessica N. and Consortium, IMAXT and Shah, Sohrab P. and Aparicio, Samuel}, journal = {Nature}, volume = {612}, number = {7938}, pages = {106--115}, year = {2022}, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s41586-022-05249-0}, }
2021
-
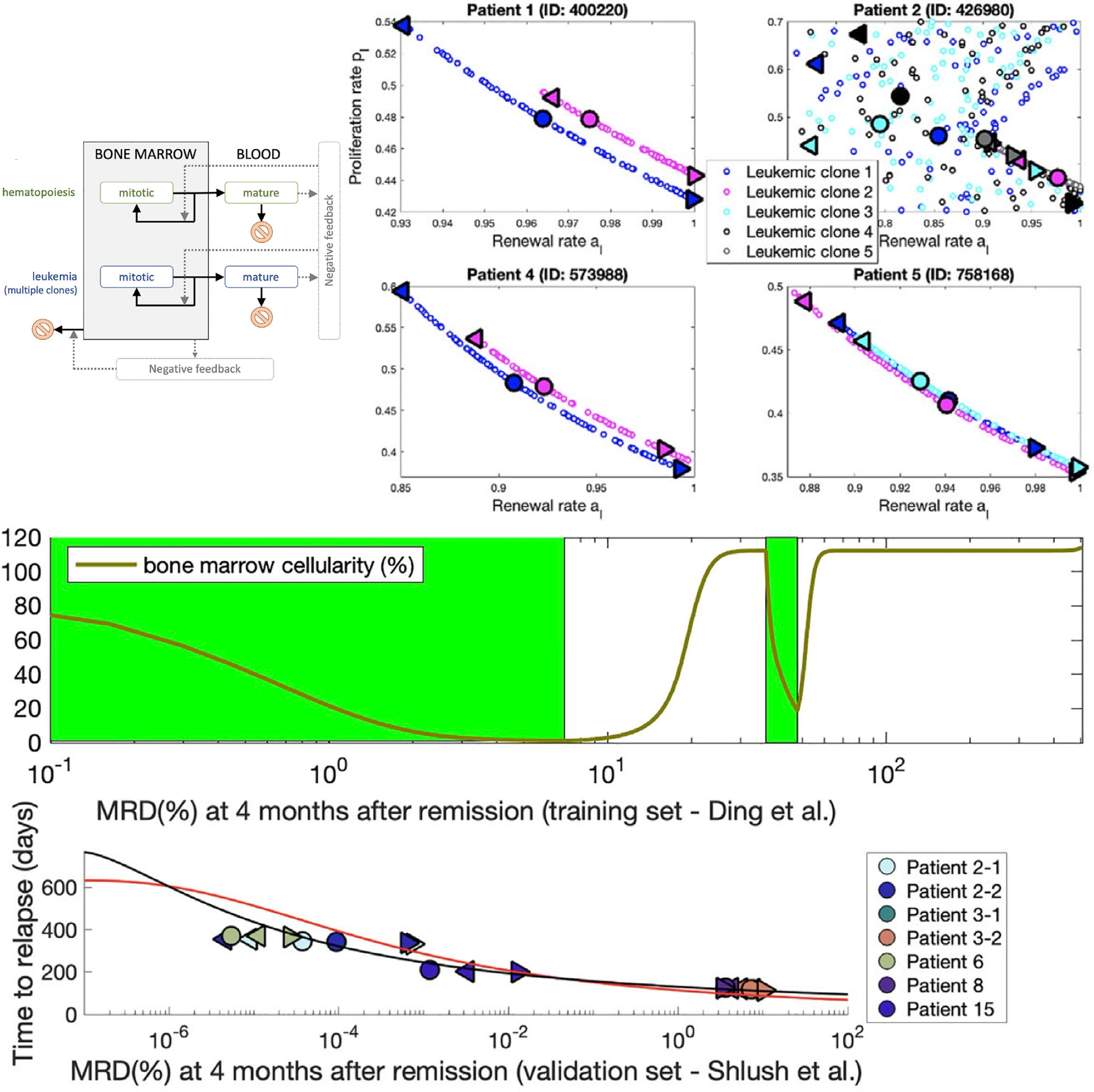 Predicting time to relapse in acute myeloid leukemia through stochastic modeling of minimal residual disease based on clonality dataComputational and systems oncology, 2021
Predicting time to relapse in acute myeloid leukemia through stochastic modeling of minimal residual disease based on clonality dataComputational and systems oncology, 2021Event-free and overall survival remain poor for patients with acute myeloid leukemia. Chemoresistant clones contributing to relapse arise from minimal residual disease (MRD) or newly acquired mutations. However, the dynamics of clones comprising MRD is poorly understood. We developed a predictive stochastic model, based on a multitype age-dependent Markov branching process, to describe how random events in MRD contribute to the heterogeneity in treatment response. We employed training and validation sets of patients who underwent whole-genome sequencing and for whom mutant clone frequencies at diagnosis and relapse were available. The disease evolution and treatment outcome are subject to stochastic fluctuations. Estimates of malignant clone growth rates, obtained by model fitting, are consistent with published data. Using the estimates from the training set, we developed a function linking MRD and time of relapse with MRD inferred from the model fits to clone frequencies and other data. An independent validation set confirmed our model. In a third dataset, we fitted the model to data at diagnosis and remission and predicted the time to relapse. As a conclusion, given bone marrow genome at diagnosis and MRD at or past remission, the model can predict time to relapse and help guide treatment decisions to mitigate relapse.
@article{dinh2021predicting, dimensions = {true}, title = {Predicting time to relapse in acute myeloid leukemia through stochastic modeling of minimal residual disease based on clonality data}, author = {Dinh, Khanh N. and Jaksik, Roman and Corey, Seth J. and Kimmel, Marek}, journal = {Computational and systems oncology}, volume = {1}, number = {3}, pages = {e1026}, year = {2021}, publisher = {Wiley Online Library}, doi = {10.1002/cso2.1026}, } -
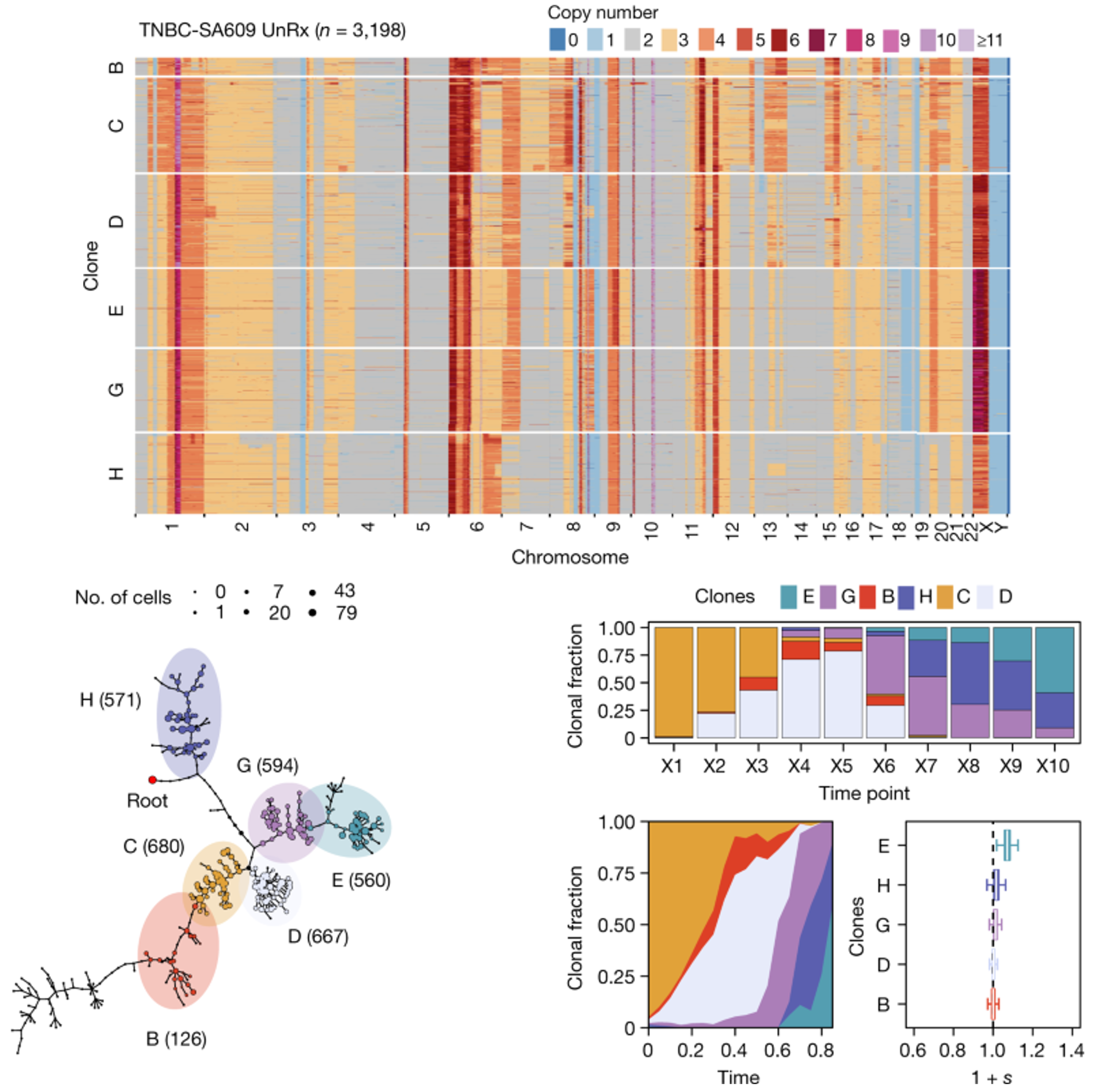 Clonal fitness inferred from time-series modelling of single-cell cancer genomesSohrab Salehi, Farhia Kabeer, Nicholas Ceglia, Mirela Andronescu, Marc J. Williams, Kieran R. Campbell, Tehmina Masud, Beixi Wang, Justina Biele, Jazmine Brimhall, David Gee, Hakwoo Lee, Jerome Ting, Allen W. Zhang, Hoa Tran, Ciara O’Flanagan, Fatemeh Dorri, Nicole Rusk, Teresa Ruiz Algara, So Ra Lee, Brian Yu Chieh Cheng, Peter Eirew, Takako Kono, Jenifer Pham, Diljot Grewal, Daniel Lai, Richard Moore, Andrew J. Mungall, Marco A. Marra, IMAXT Consortium, Andrew McPherson, Alexandre Bouchard-Côté, Samuel Aparicio, and Sohrab P. ShahNature, 2021
Clonal fitness inferred from time-series modelling of single-cell cancer genomesSohrab Salehi, Farhia Kabeer, Nicholas Ceglia, Mirela Andronescu, Marc J. Williams, Kieran R. Campbell, Tehmina Masud, Beixi Wang, Justina Biele, Jazmine Brimhall, David Gee, Hakwoo Lee, Jerome Ting, Allen W. Zhang, Hoa Tran, Ciara O’Flanagan, Fatemeh Dorri, Nicole Rusk, Teresa Ruiz Algara, So Ra Lee, Brian Yu Chieh Cheng, Peter Eirew, Takako Kono, Jenifer Pham, Diljot Grewal, Daniel Lai, Richard Moore, Andrew J. Mungall, Marco A. Marra, IMAXT Consortium, Andrew McPherson, Alexandre Bouchard-Côté, Samuel Aparicio, and Sohrab P. ShahNature, 2021Progress in defining genomic fitness landscapes in cancer, especially those defined by copy number alterations (CNAs), has been impeded by lack of time-series single-cell sampling of polyclonal populations and temporal statistical models. Here we generated 42,000 genomes from multi-year time-series single-cell whole-genome sequencing of breast epithelium and primary triple-negative breast cancer (TNBC) patient-derived xenografts (PDXs), revealing the nature of CNA-defined clonal fitness dynamics induced by TP53 mutation and cisplatin chemotherapy. Using a new Wright-Fisher population genetics model to infer clonal fitness, we found that TP53 mutation alters the fitness landscape, reproducibly distributing fitness over a larger number of clones associated with distinct CNAs. Furthermore, in TNBC PDX models with mutated TP53, inferred fitness coefficients from CNA-based genotypes accurately forecast experimentally enforced clonal competition dynamics. Drug treatment in three long-term serially passaged TNBC PDXs resulted in cisplatin-resistant clones emerging from low-fitness phylogenetic lineages in the untreated setting. Conversely, high-fitness clones from treatment-naive controls were eradicated, signalling an inversion of the fitness landscape. Finally, upon release of drug, selection pressure dynamics were reversed, indicating a fitness cost of treatment resistance. Together, our findings define clonal fitness linked to both CNA and therapeutic resistance in polyclonal tumours.
@article{salehi2021clonal, dimensions = {true}, title = {Clonal fitness inferred from time-series modelling of single-cell cancer genomes}, author = {Salehi, Sohrab and Kabeer, Farhia and Ceglia, Nicholas and Andronescu, Mirela and Williams, Marc J. and Campbell, Kieran R. and Masud, Tehmina and Wang, Beixi and Biele, Justina and Brimhall, Jazmine and Gee, David and Lee, Hakwoo and Ting, Jerome and Zhang, Allen W. and Tran, Hoa and O'Flanagan, Ciara and Dorri, Fatemeh and Rusk, Nicole and de Algara, Teresa Ruiz and Lee, So Ra and Cheng, Brian Yu Chieh and Eirew, Peter and Kono, Takako and Pham, Jenifer and Grewal, Diljot and Lai, Daniel and Moore, Richard and Mungall, Andrew J. and Marra, Marco A. and Consortium, IMAXT and McPherson, Andrew and Bouchard-C{\o}t{\'e}, Alexandre and Aparicio, Samuel and Shah, Sohrab P.}, journal = {Nature}, volume = {595}, number = {7868}, pages = {585--590}, year = {2021}, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s41586-021-03648-3}, } -
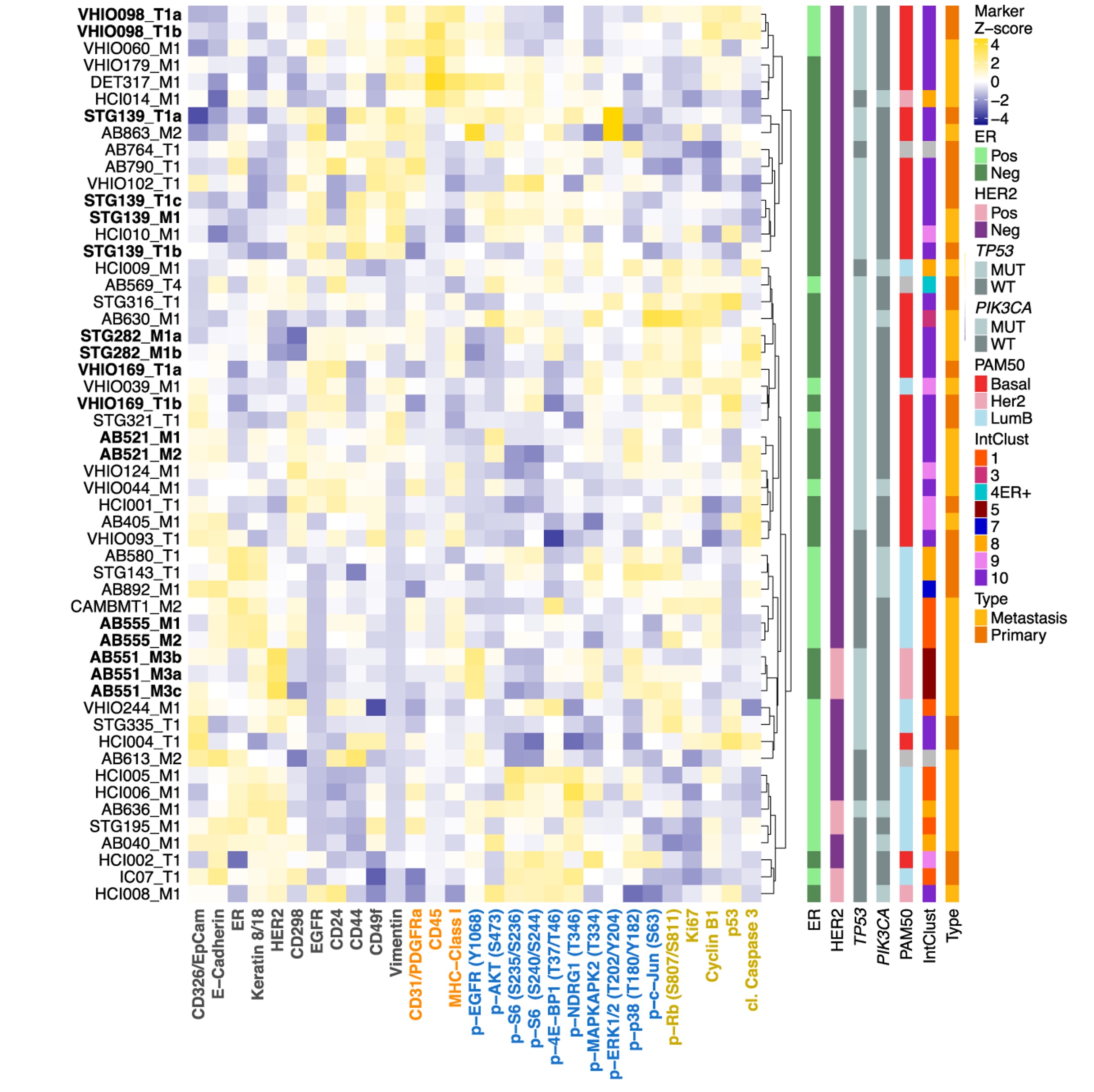 Landscapes of cellular phenotypic diversity in breast cancer xenografts and their impact on drug responseDimitra Georgopoulou, Maurizio Callari, Oscar M. Rueda, Abigail Shea, Alistair Martin, Agnese Giovannetti, Fatime Qosaj, Ali Dariush, Suet-Feung Chin, Larissa S. Carnevalli, Elena Provenzano, Wendy Greenwood, Giulia Lerda, Elham Esmaeilishirazifard, Martin O’Reilly, Violeta Serra, Dario Bressan, IMAXT Consortium, Gordon B. Mills, H. Raza Ali, Sabina S. Cosulich, Gregory J. Hannon, Alejandra Bruna, and Carlos CaldasNature Communications, 2021
Landscapes of cellular phenotypic diversity in breast cancer xenografts and their impact on drug responseDimitra Georgopoulou, Maurizio Callari, Oscar M. Rueda, Abigail Shea, Alistair Martin, Agnese Giovannetti, Fatime Qosaj, Ali Dariush, Suet-Feung Chin, Larissa S. Carnevalli, Elena Provenzano, Wendy Greenwood, Giulia Lerda, Elham Esmaeilishirazifard, Martin O’Reilly, Violeta Serra, Dario Bressan, IMAXT Consortium, Gordon B. Mills, H. Raza Ali, Sabina S. Cosulich, Gregory J. Hannon, Alejandra Bruna, and Carlos CaldasNature Communications, 2021The heterogeneity of breast cancer plays a major role in drug response and resistance and has been extensively characterized at the genomic level. Here, a single-cell breast cancer mass cytometry (BCMC) panel is optimized to identify cell phenotypes and their oncogenic signalling states in a biobank of patient-derived tumour xenograft (PDTX) models representing the diversity of human breast cancer. The BCMC panel identifies 13 cellular phenotypes (11 human and 2 murine), associated with both breast cancer subtypes and specific genomic features. Pre-treatment cellular phenotypic composition is a determinant of response to anticancer therapies. Single-cell profiling also reveals drug-induced cellular phenotypic dynamics, unravelling previously unnoticed intra-tumour response diversity. The comprehensive view of the landscapes of cellular phenotypic heterogeneity in PDTXs uncovered by the BCMC panel, which is mirrored in primary human tumours, has profound implications for understanding and predicting therapy response and resistance.
@article{georgopoulou2021landscapes, dimensions = {true}, title = {Landscapes of cellular phenotypic diversity in breast cancer xenografts and their impact on drug response}, author = {Georgopoulou, Dimitra and Callari, Maurizio and Rueda, Oscar M. and Shea, Abigail and Martin, Alistair and Giovannetti, Agnese and Qosaj, Fatime and Dariush, Ali and Chin, Suet-Feung and Carnevalli, Larissa S. and Provenzano, Elena and Greenwood, Wendy and Lerda, Giulia and Esmaeilishirazifard, Elham and O'Reilly, Martin and Serra, Violeta and Bressan, Dario and Consortium, IMAXT and Mills, Gordon B. and Ali, H. Raza and Cosulich, Sabina S. and Hannon, Gregory J. and Bruna, Alejandra and Caldas, Carlos}, journal = {Nature Communications}, volume = {12}, number = {1}, pages = {1998}, year = {2021}, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s41467-021-22303-z}, }
2020
-
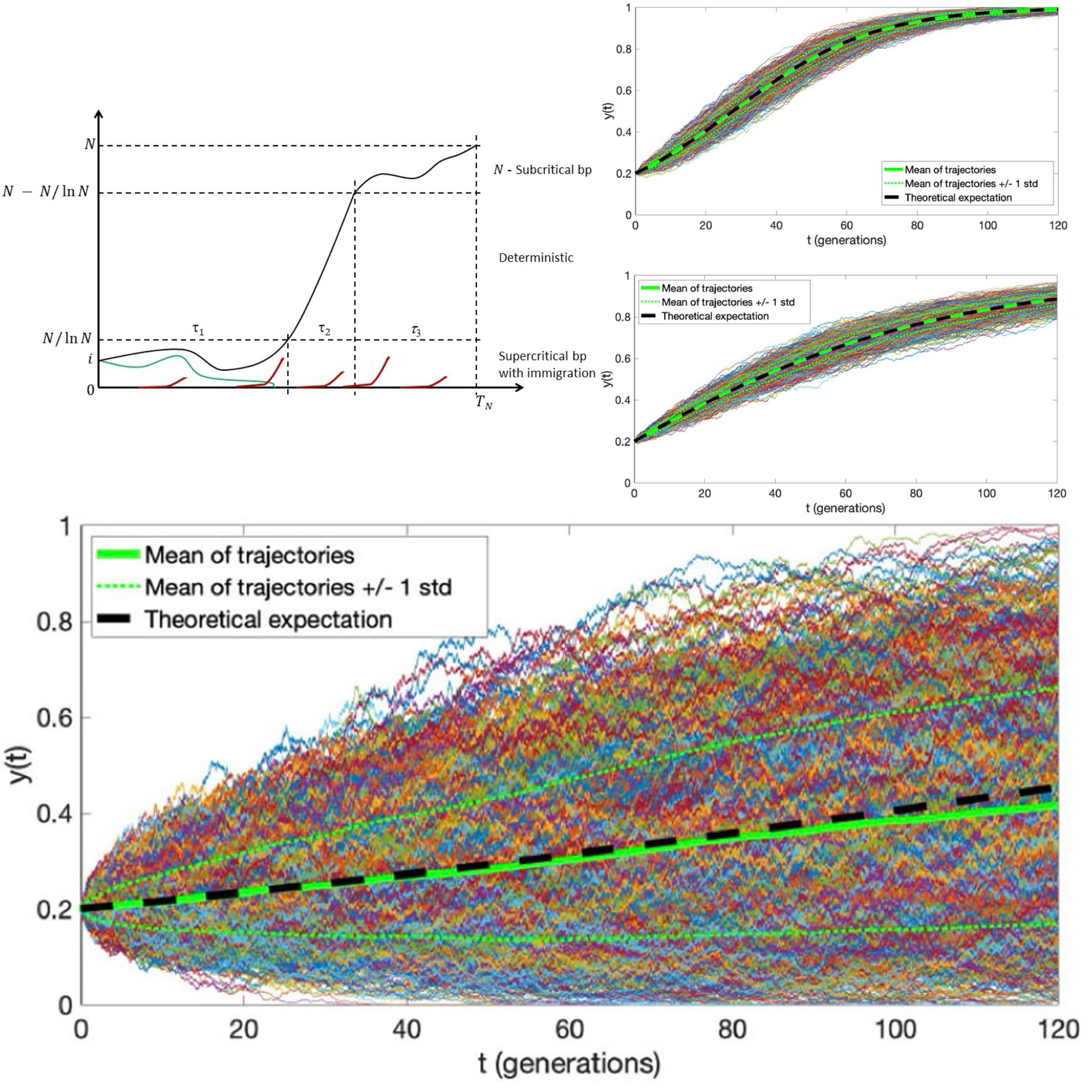 Application of the Moran model in estimating selection coefficient of mutated CSF3R clones in the evolution of severe congenital neutropenia to myeloid neoplasiaKhanh N. Dinh, Seth J. Corey, and Marek KimmelFrontiers in Physiology, 2020
Application of the Moran model in estimating selection coefficient of mutated CSF3R clones in the evolution of severe congenital neutropenia to myeloid neoplasiaKhanh N. Dinh, Seth J. Corey, and Marek KimmelFrontiers in Physiology, 2020Bone marrow failure (BMF) syndromes, such as severe congenital neutropenia (SCN) are leukemia predisposition syndromes. We focus here on the transition from SCN to pre-leukemic myelodysplastic syndrome (MDS). Stochastic mathematical models have been conceived that attempt to explain the transition of SCN to MDS, in the most parsimonious way, using extensions of standard processes of population genetics and population dynamics, such as the branching and the Moran processes. We previously presented a hypothesis of the SCN to MDS transition, which involves directional selection and recurrent mutation, to explain the distribution of ages at onset of MDS or AML. Based on experimental and clinical data and a model of human hematopoiesis, a range of probable values of the selection coefficient s and mutation rate μ have been determined. These estimates lead to predictions of the age at onset of MDS or AML, which are consistent with the clinical data. In the current paper, based on data extracted from published literature, we seek to provide an independent validation of these estimates. We proceed with two purposes in mind: (i) to determine the ballpark estimates of the selection coefficients and verify their consistency with those previously obtained and (ii) to provide possible insight into the role of recurrent mutations of the G-CSF receptor in the SCN to MDS transition.
@article{dinh2020application, dimensions = {true}, title = {Application of the Moran model in estimating selection coefficient of mutated CSF3R clones in the evolution of severe congenital neutropenia to myeloid neoplasia}, author = {Dinh, Khanh N. and Corey, Seth J. and Kimmel, Marek}, journal = {Frontiers in Physiology}, volume = {11}, pages = {806}, year = {2020}, publisher = {Frontiers Media SA}, doi = {10.3389/fphys.2020.00806}, } -
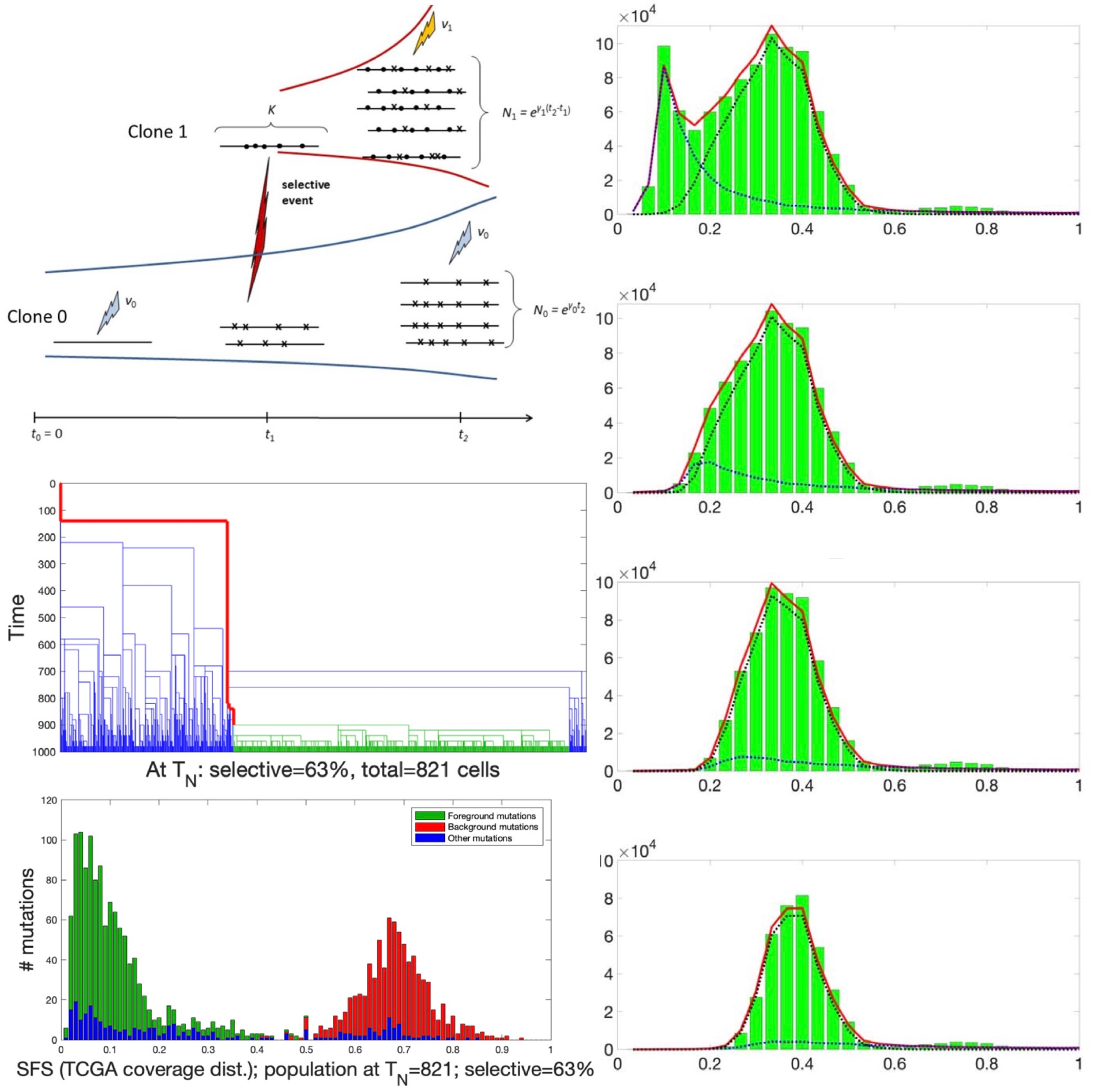 Statistical inference for the evolutionary history of cancer genomesStatistical Science, 2020
Statistical inference for the evolutionary history of cancer genomesStatistical Science, 2020Recent years have seen considerable work on inference about cancer evolution from mutations identified in cancer samples. Much of the modeling work has been based on classical models of population genetics, generalized to accommodate time-varying cell population size. Reverse-time, genealogical views of such models, commonly known as coalescents, have been used to infer aspects of the past of growing populations. Another approach is to use branching processes, the simplest scenario being the classical linear birth-death process. Inference from evolutionary models of DNA often exploits summary statistics of the sequence data, a common one being the so-called Site Frequency Spectrum (SFS). In a bulk tumor sequencing experiment, we can estimate for each site at which a novel somatic point mutation has arisen, the proportion of cells that carry that mutation. These numbers are then grouped into collections of sites which have similar mutant fractions. We examine how the SFS based on birth-death processes differs from those based on the coalescent model. This may stem from the different sampling mechanisms in the two approaches. However, we also show that despite this, they are quantitatively comparable for the range of parameters typical for tumor cell populations. We also present a model of tumor evolution with selective sweeps, and demonstrate how it may help in understanding the history of a tumor as well as the influence of data pre-processing. We illustrate the theory with applications to several examples from The Cancer Genome Atlas tumors.
@article{dinh2020statistical, dimensions = {true}, title = {Statistical inference for the evolutionary history of cancer genomes}, author = {Dinh, Khanh N. and Jaksik, Roman and Kimmel, Marek and Lambert, Amaury and Tavar{\'e}, Simon}, journal = {Statistical Science}, volume = {35}, number = {1}, pages = {129--144}, year = {2020}, publisher = {JSTOR}, doi = {10.1214/19-sts7561}, }
2019
-
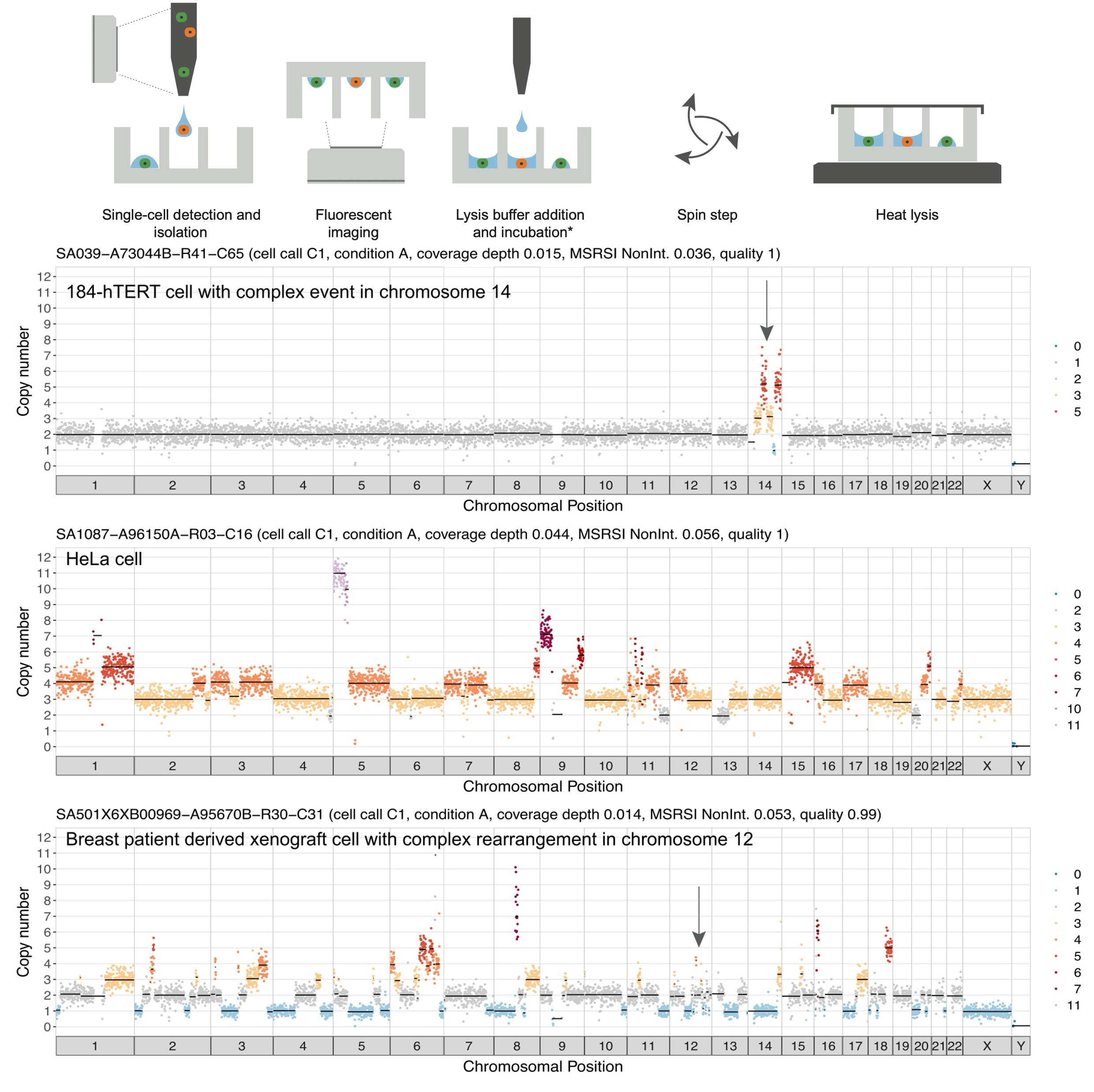 Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencingEmma Laks, Andrew McPherson, Hans Zahn, Daniel Lai, Adi Steif, Jazmine Brimhall, Justina Biele, Beixi Wang, Tehmina Masud, Jerome Ting, Diljot Grewal, Cydney Nielsen, Samantha Leung, Viktoria Bojilova, Maia Smith, Oleg Golovko, Steven Poon, Peter Eirew, Farhia Kabeer, Teresa Algara, So Ra Lee, M. Jafar Taghiyar, Curtis Huebner, Jessica Ngo , Tim Chan, Spencer Vatrt-Watts, Pascale Walters, Nafis Abrar , Sophia Chan, Matt Wiens, Lauren Martin, R. Wilder Scott, T. Michael Underhill, Elizabeth Chavez, Christian Steidl, Daniel Da Costa, Yussanne Ma, Robin J. N. Coope, Richard Corbett, Stephen Pleasance, Richard Moore, Andrew J. Mungall, Colin Mar, Fergus Cafferty, Karen Gelmon, Stephen Chia, IMAXT Consortium, Marco A. Marra, Carl Hansen, Sohrab P. Shah, and Samuel AparicioCell, 2019
Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencingEmma Laks, Andrew McPherson, Hans Zahn, Daniel Lai, Adi Steif, Jazmine Brimhall, Justina Biele, Beixi Wang, Tehmina Masud, Jerome Ting, Diljot Grewal, Cydney Nielsen, Samantha Leung, Viktoria Bojilova, Maia Smith, Oleg Golovko, Steven Poon, Peter Eirew, Farhia Kabeer, Teresa Algara, So Ra Lee, M. Jafar Taghiyar, Curtis Huebner, Jessica Ngo , Tim Chan, Spencer Vatrt-Watts, Pascale Walters, Nafis Abrar , Sophia Chan, Matt Wiens, Lauren Martin, R. Wilder Scott, T. Michael Underhill, Elizabeth Chavez, Christian Steidl, Daniel Da Costa, Yussanne Ma, Robin J. N. Coope, Richard Corbett, Stephen Pleasance, Richard Moore, Andrew J. Mungall, Colin Mar, Fergus Cafferty, Karen Gelmon, Stephen Chia, IMAXT Consortium, Marco A. Marra, Carl Hansen, Sohrab P. Shah, and Samuel AparicioCell, 2019Accurate measurement of clonal genotypes, mutational processes, and replication states from individual tumor-cell genomes will facilitate improved understanding of tumor evolution. We have developed DLP+, a scalable single-cell whole-genome sequencing platform implemented using commodity instruments, image-based object recognition, and open source computational methods. Using DLP+, we have generated a resource of 51,926 single-cell genomes and matched cell images from diverse cell types including cell lines, xenografts, and diagnostic samples with limited material. From this resource we have defined variation in mitotic mis-segregation rates across tissue types and genotypes. Analysis of matched genomic and image measurements revealed correlations between cellular morphology and genome ploidy states. Aggregation of cells sharing copy number profiles allowed for calculation of single-nucleotide resolution clonal genotypes and inference of clonal phylogenies and avoided the limitations of bulk deconvolution. Finally, joint analysis over the above features defined clone-specific chromosomal aneuploidy in polyclonal populations.
@article{laks2019clonal, dimensions = {true}, title = {Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencing}, author = {Laks, Emma and McPherson, Andrew and Zahn, Hans and Lai, Daniel and Steif, Adi and Brimhall, Jazmine and Biele, Justina and Wang, Beixi and Masud, Tehmina and Ting, Jerome and Grewal, Diljot and Nielsen, Cydney and Leung, Samantha and Bojilova, Viktoria and Smith, Maia and Golovko, Oleg and Poon, Steven and Eirew, Peter and Kabeer, Farhia and Ruiz de Algara, Teresa and Lee, So Ra and Taghiyar, M. Jafar and Huebner, Curtis and Ngo, Jessica and Chan, Tim and Vatrt-Watts, Spencer and Walters, Pascale and Abrar, Nafis and Chan, Sophia and Wiens, Matt and Martin, Lauren and Scott, R. Wilder and Underhill, T. Michael and Chavez, Elizabeth and Steidl, Christian and Da Costa, Daniel and Ma, Yussanne and Coope, Robin J. N. and Corbett, Richard and Pleasance, Stephen and Moore, Richard and Mungall, Andrew J. and Mar, Colin and Cafferty, Fergus and Gelmon, Karen and Chia, Stephen and Consortium, IMAXT and Marra, Marco A. and Hansen, Carl and Shah, Sohrab P. and Aparicio, Samuel}, journal = {Cell}, volume = {179}, number = {5}, pages = {1207--1221}, year = {2019}, publisher = {Elsevier}, doi = {10.1016/j.cell.2019.10.026}, }
2018
-
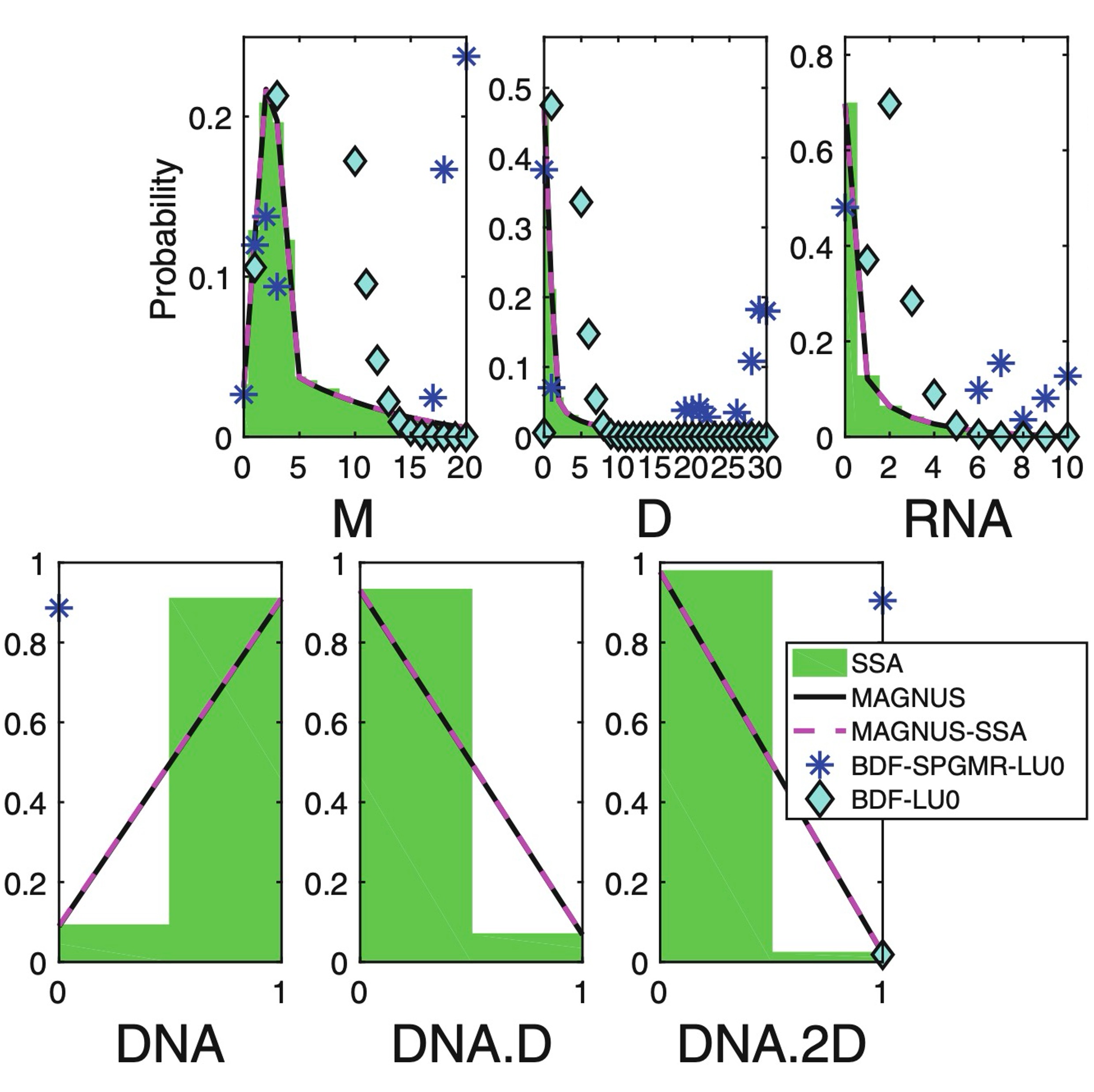 A comparison of the Magnus expansion and other solvers for the chemical master equation with variable ratesKhanh N. Dinh, and Roger B. SidjeIn Recent Advances in Mathematical and Statistical Methods: IV AMMCS International Conference, Waterloo, Canada, August 20–25, 2017 IV, 2018
A comparison of the Magnus expansion and other solvers for the chemical master equation with variable ratesKhanh N. Dinh, and Roger B. SidjeIn Recent Advances in Mathematical and Statistical Methods: IV AMMCS International Conference, Waterloo, Canada, August 20–25, 2017 IV, 2018Many traditional approaches for solving the chemical master equation (CME) cannot be used in their basic form when reaction rates change over time, for instance due to cell volume or temperature. One technique is to use the Magnus expansion to represent the solution to the CME as the action of a matrix exponential, for which Krylov-based approximation methods can be applied. In this paper, we compare two variants of the Magnus scheme with some popular ordinary differential equations (ODE) solvers, such as Adams-Bashforth, Runge-Kutta and Backward-differentiation formula (BDF). Our numerical tests show that the Magnus variants are remarkably efficient at computing the transient probability distributions of a transcriptional regulatory system where propensities vary over time due to cell volume increase.
@inproceedings{dinh2018comparison, dimensions = {true}, title = {A comparison of the Magnus expansion and other solvers for the chemical master equation with variable rates}, author = {Dinh, Khanh N. and Sidje, Roger B.}, booktitle = {Recent Advances in Mathematical and Statistical Methods: IV AMMCS International Conference, Waterloo, Canada, August 20--25, 2017 IV}, pages = {261--270}, year = {2018}, organization = {Springer}, doi = {10.1007/978-3-319-99719-3_24}, } -
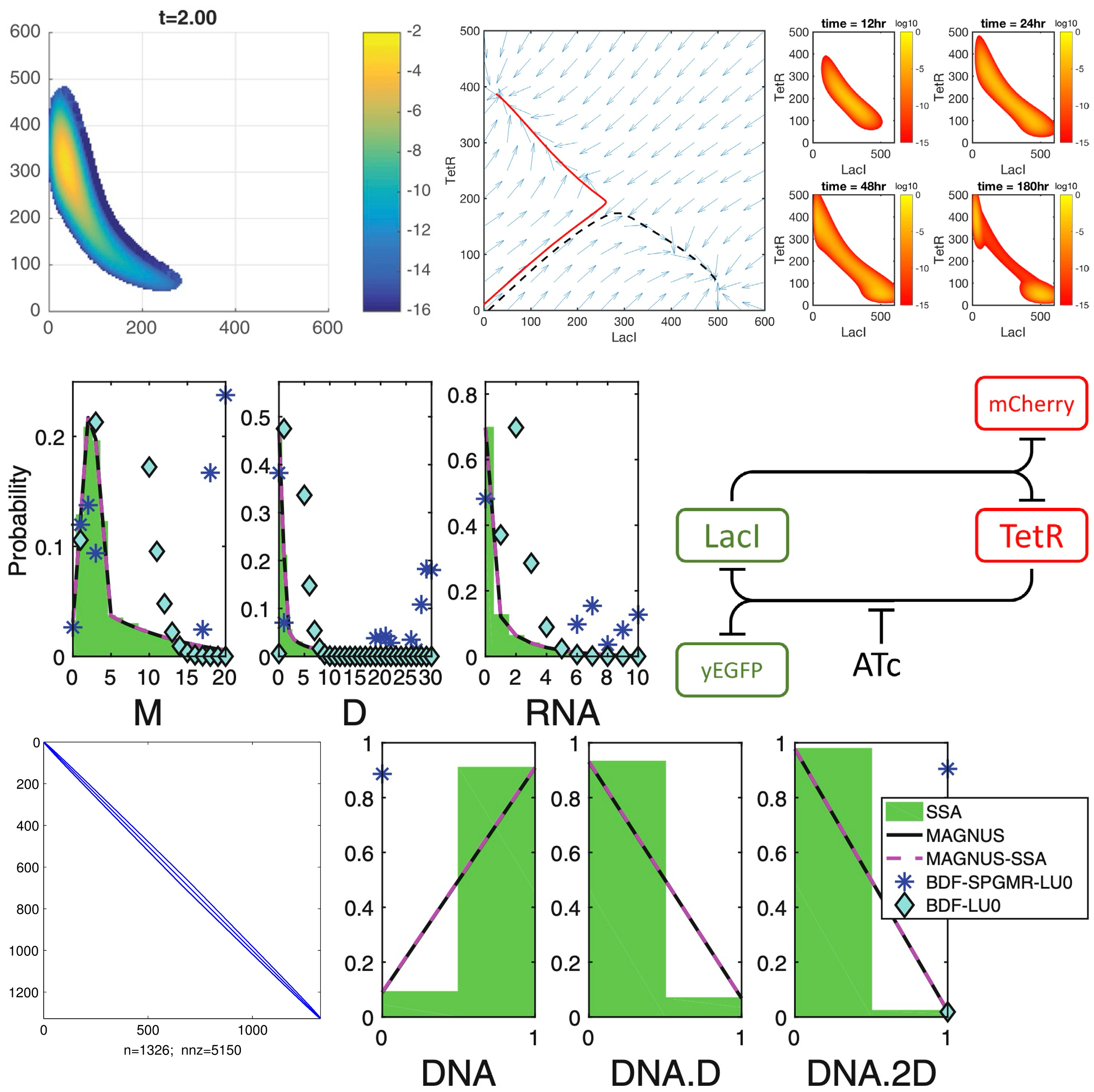 Inexact methods for the chemical master equation with constant or time-varying propensities, and application to parameter inferenceKhanh N. DinhDoctoral thesis , 2018
Inexact methods for the chemical master equation with constant or time-varying propensities, and application to parameter inferenceKhanh N. DinhDoctoral thesis , 2018Complex reaction networks arise in molecular biology and many other different fields of science such as ecology and social study. A familiar approach to modeling such problems is to find their master equation. In systems biology, the equation is called the chemical master equation (CME), and solving the CME is a difficult task, because of the curse of dimensionality. The goal of this dissertation is to alleviate this curse via the use of the finite state projection (FSP), in both cases where the CME matrix is constant (if the reaction rates are time-independent) or time-varying (if the reaction rates change over time). The work includes a theoretical characterization of the FSP truncation technique by showing that it can be put in the framework of inexact Krylov methods that relax matrix-vector products and compute them expediently by trading accuracy for speed. We also examine practical applications of our work in delay CME and parameter inference through local and global optimization schemes.
@book{dinh2018inexact, dimensions = {true}, title = {Inexact methods for the chemical master equation with constant or time-varying propensities, and application to parameter inference}, author = {Dinh, Khanh N.}, journal = {The University of Alabama}, year = {2018}, }
2017
-
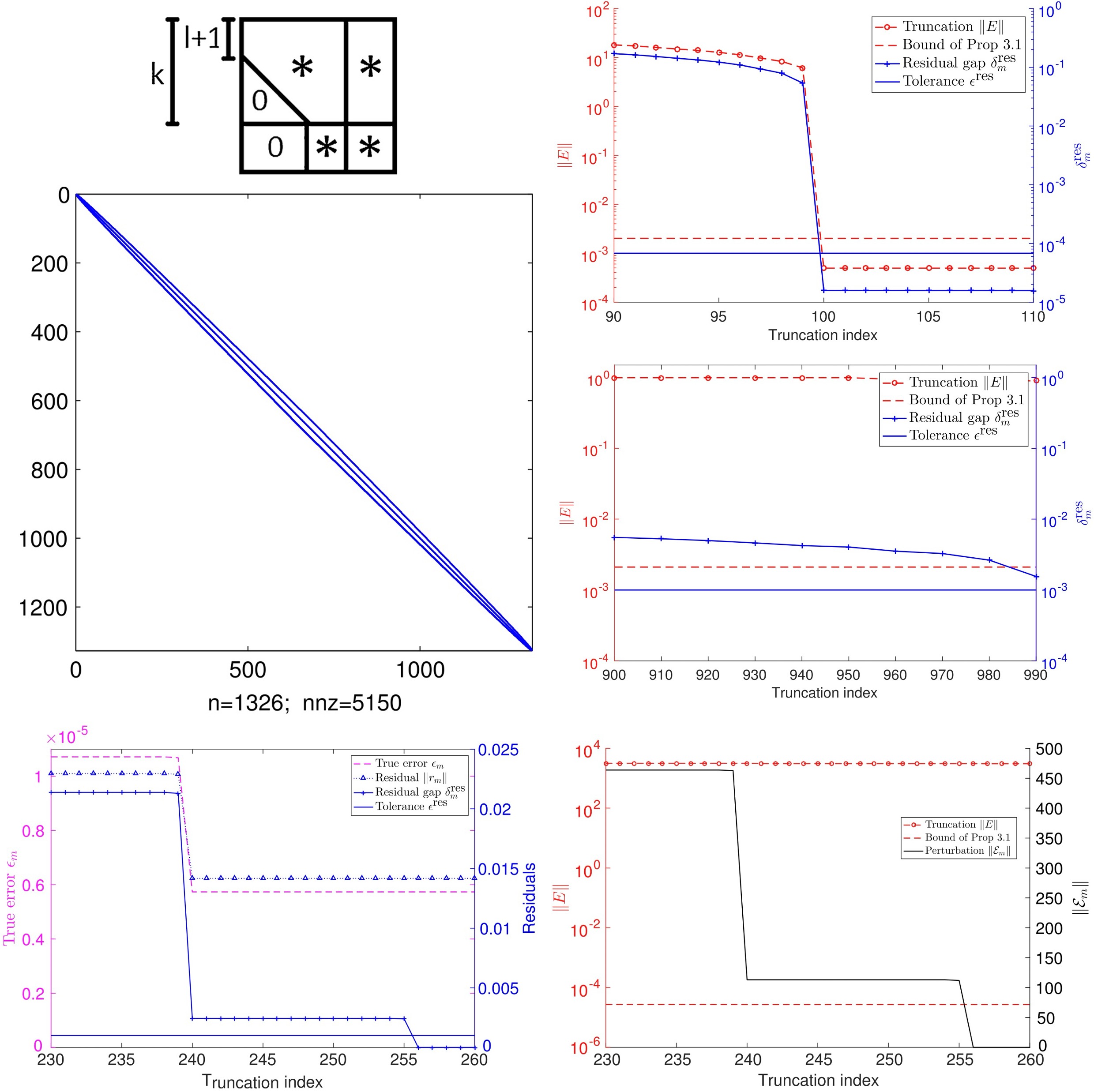 Analysis of inexact Krylov subspace methods for approximating the matrix exponentialKhanh N. Dinh, and Roger B. SidjeMathematics and Computers in Simulation, 2017
Analysis of inexact Krylov subspace methods for approximating the matrix exponentialKhanh N. Dinh, and Roger B. SidjeMathematics and Computers in Simulation, 2017Krylov subspace methods have proved quite effective at approximating the action of a large sparse matrix exponential on a vector. Their numerical robustness and matrix-free nature have enabled them to make inroads into a variety of applications. A case in point is solving the chemical master equation (CME) in the context of modeling biochemical reactions in biological cells. This is a challenging problem that gives rise to an extremely large matrix due to the curse of dimensionality. Inexact Krylov subspace methods that build on truncation techniques have helped solve some CME models that were considered computationally out of reach as recently as a few years ago. However, as models grow, truncating them means using an even smaller fraction of their whole extent, thereby introducing more inexactness. But experimental evidence suggests an apparent success and the aim of this study is to give theoretical insights into the reasons why. Essentially, we show that the truncation can be put in the framework of inexact Krylov methods that relax matrix–vector products and compute them expediently by trading accuracy for speed. This allows us to analyze both the residual (or defect) and the error of the resulting approximations to the matrix exponential from the viewpoint of inexact Krylov methods.
@article{dinh2017analysis, dimensions = {true}, title = {Analysis of inexact Krylov subspace methods for approximating the matrix exponential}, author = {Dinh, Khanh N. and Sidje, Roger B.}, journal = {Mathematics and Computers in Simulation}, volume = {138}, pages = {1--13}, year = {2017}, publisher = {Elsevier}, doi = {10.1016/j.matcom.2017.01.002}, } -
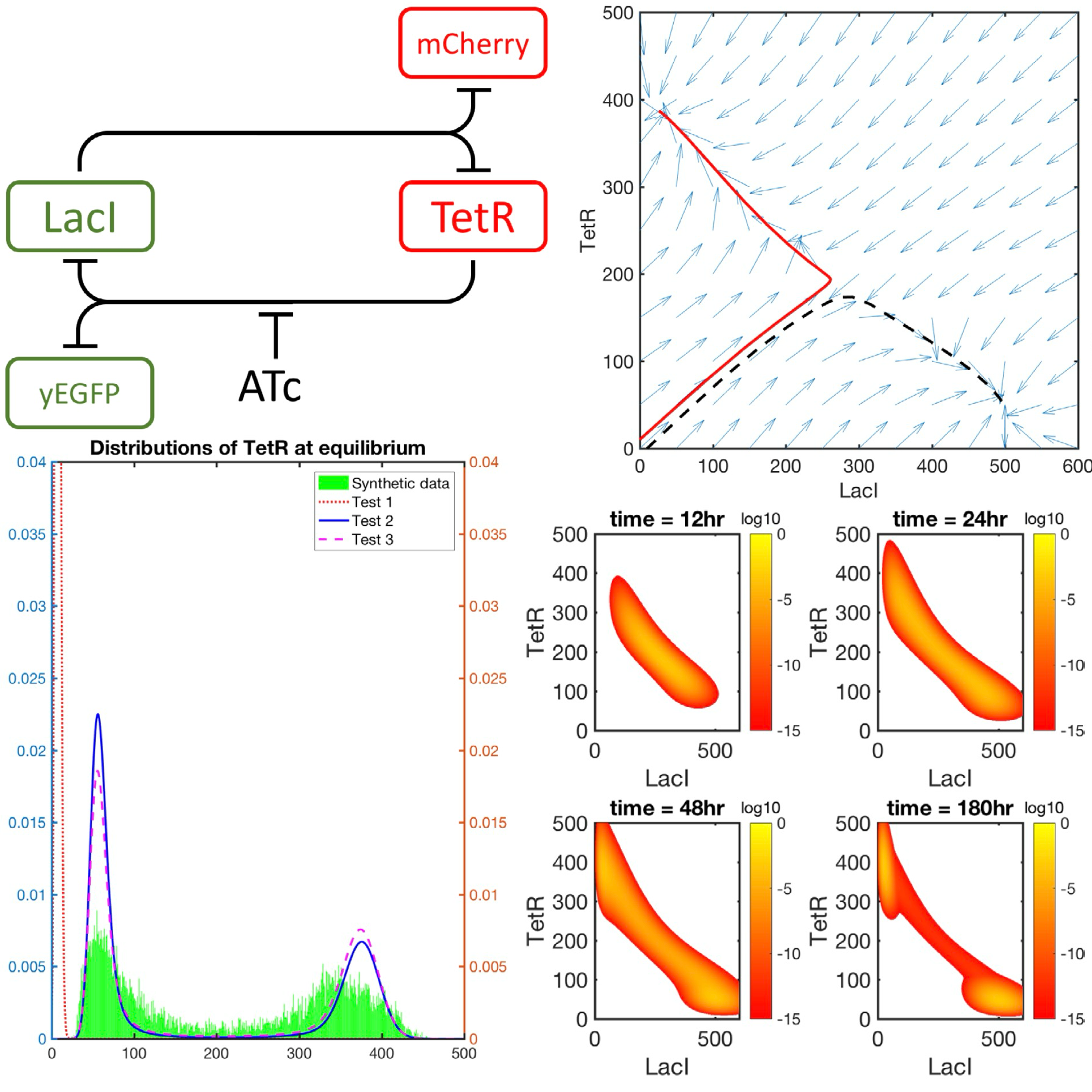 An application of the Krylov-FSP-SSA method to parameter fitting with maximum likelihoodKhanh N. Dinh, and Roger B. SidjePhysical Biology, 2017
An application of the Krylov-FSP-SSA method to parameter fitting with maximum likelihoodKhanh N. Dinh, and Roger B. SidjePhysical Biology, 2017Monte Carlo methods such as the stochastic simulation algorithm (SSA) have traditionally been employed in gene regulation problems. However, there has been increasing interest to directly obtain the probability distribution of the molecules involved by solving the chemical master equation (CME). This requires addressing the curse of dimensionality that is inherent in most gene regulation problems. The finite state projection (FSP) seeks to address the challenge and there have been variants that further reduce the size of the projection or that accelerate the resulting matrix exponential. The Krylov-FSP-SSA variant has proved numerically efficient by combining, on one hand, the SSA to adaptively drive the FSP, and on the other hand, adaptive Krylov techniques to evaluate the matrix exponential. Here we apply this Krylov-FSP-SSA to a mutual inhibitory gene network synthetically engineered in Saccharomyces cerevisiae, in which bimodality arises. We show numerically that the approach can efficiently approximate the transient probability distribution, and this has important implications for parameter fitting, where the CME has to be solved for many different parameter sets. The fitting scheme amounts to an optimization problem of finding the parameter set so that the transient probability distributions fit the observations with maximum likelihood. We compare five optimization schemes for this difficult problem, thereby providing further insights into this approach of parameter estimation that is often applied to models in systems biology where there is a need to calibrate free parameters.
@article{dinh2017application, dimensions = {true}, title = {An application of the Krylov-FSP-SSA method to parameter fitting with maximum likelihood}, author = {Dinh, Khanh N. and Sidje, Roger B.}, journal = {Physical Biology}, volume = {14}, number = {6}, pages = {065001}, year = {2017}, publisher = {IOP Publishing}, doi = {10.1088/1478-3975/aa868a}, } -
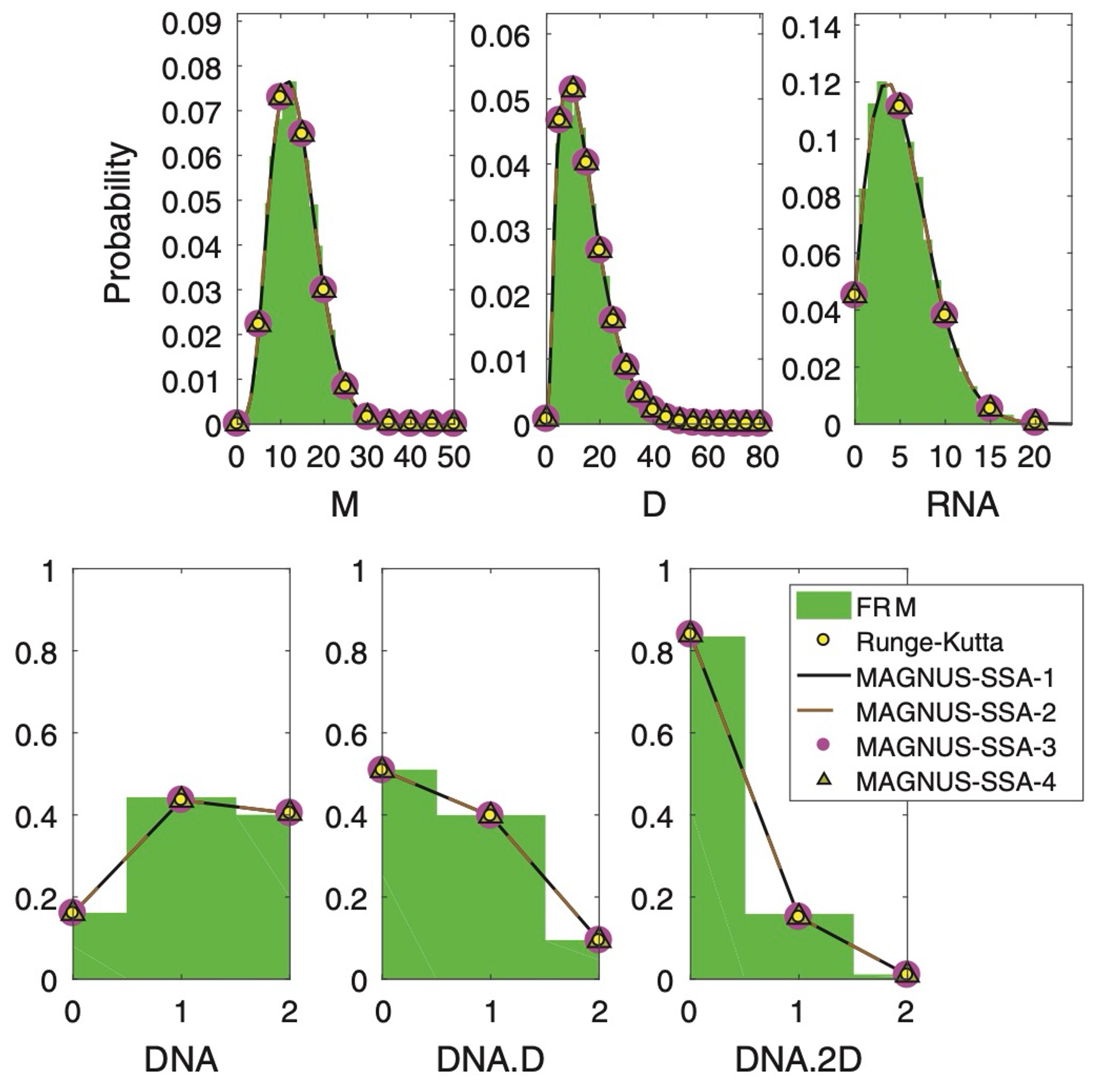 An adaptive Magnus expansion method for solving the chemical master equation with time-dependent propensitiesKhanh N. Dinh, and Roger B. SidjeJournal of Coupled Systems and Multiscale Dynamics, 2017
An adaptive Magnus expansion method for solving the chemical master equation with time-dependent propensitiesKhanh N. Dinh, and Roger B. SidjeJournal of Coupled Systems and Multiscale Dynamics, 2017The chemical master equation (CME) is a system of ordinary differential equations (ODEs) to model the chemical interaction of molecular species. The largeness of the state space of the system makes solving the CME difficult, and this has motivated reduction strategies such as the finite state projection (FSP). Moreover, if the reaction rates are functions of the time, the CME becomes an ODE problem with time-dependent coefficients. Solution techniques include Monte Carlo algorithms, such as the stochastic simulation algorithm (SSA) or ODE solvers, such as Adams-PECE, Runge-Kutta and backward-differentiation formula (BDF). There are also Magnus-based solvers that have however not been thoroughly explored in the CME context. Here we introduce an adaptive time-stepping Magnus-SSA algorithm, in which the CME is solved using a Magnus expansion with not only a variable time-step but also with a variable state space that changes at each step via the SSA, and several error approximation approaches are attempted to monitor the adaptivity. We perform comparative tests against the classical Adams-PECE, Runge-Kutta and BDF methods on three biological problems, showing that the proposed adaptive Magnus-based variants can be efficient when the CME with time-dependent rates is stiff.
@article{dinh2017adaptive, dimensions = {true}, title = {An adaptive Magnus expansion method for solving the chemical master equation with time-dependent propensities}, author = {Dinh, Khanh N. and Sidje, Roger B.}, journal = {Journal of Coupled Systems and Multiscale Dynamics}, volume = {5}, number = {2}, pages = {119--131}, year = {2017}, publisher = {American Scientific Publishers}, doi = {10.1166/jcsmd.2017.1124}, }
2016
-
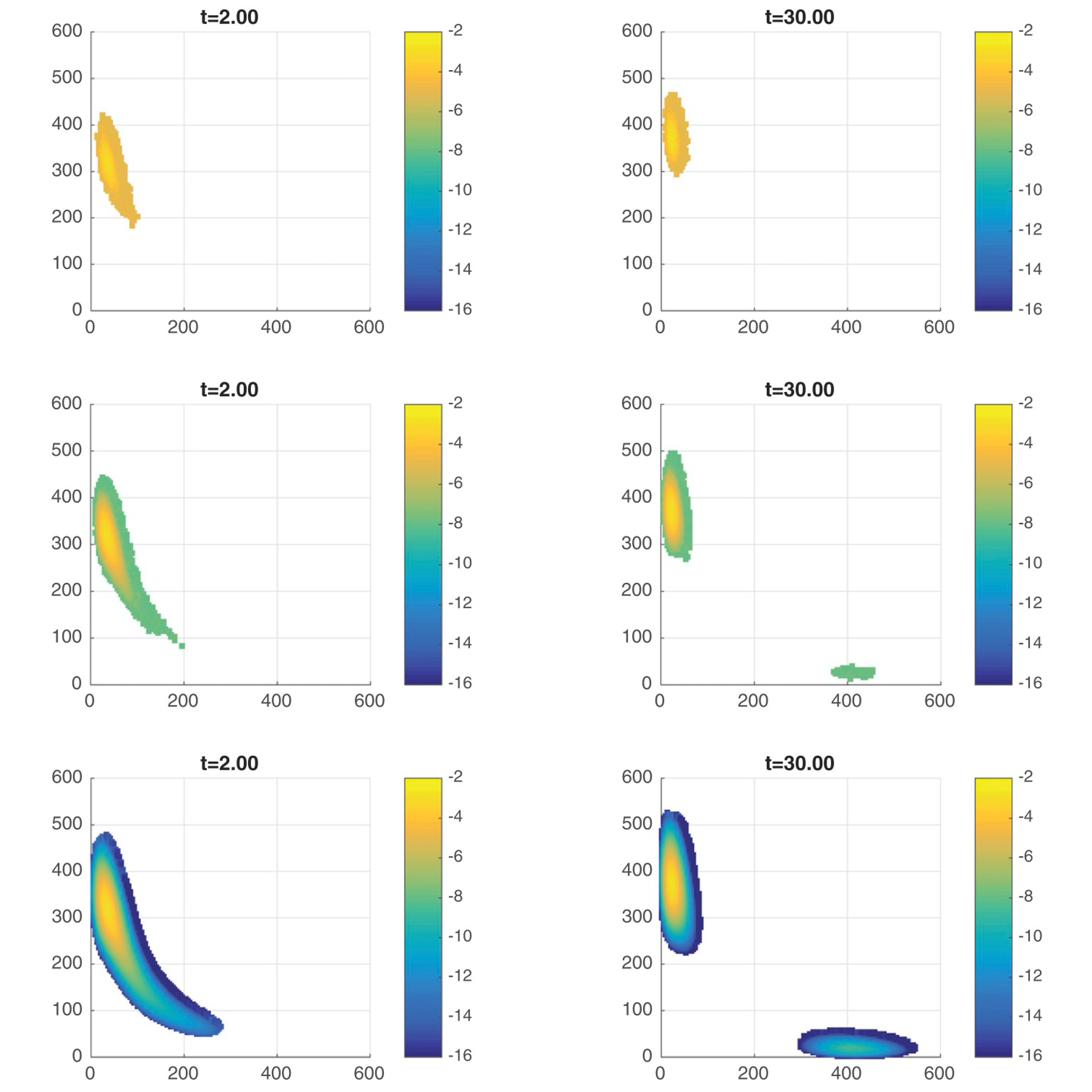 Understanding the finite state projection and related methods for solving the chemical master equationKhanh N. Dinh, and Roger B. SidjePhysical Biology, 2016
Understanding the finite state projection and related methods for solving the chemical master equationKhanh N. Dinh, and Roger B. SidjePhysical Biology, 2016The finite state projection (FSP) method has enabled us to solve the chemical master equation of some biological models that were considered out of reach not long ago. Since the original FSP method, much effort has gone into transforming it into an adaptive time-stepping algorithm as well as studying its accuracy. Some of the improvements include the multiple time interval FSP, the sliding windows, and most notably the Krylov-FSP approach. Our goal in this tutorial is to give the reader an overview of the current methods that build on the FSP.
@article{dinh2016understanding, dimensions = {true}, title = {Understanding the finite state projection and related methods for solving the chemical master equation}, author = {Dinh, Khanh N. and Sidje, Roger B.}, journal = {Physical Biology}, volume = {13}, number = {3}, pages = {035003}, year = {2016}, publisher = {IOP Publishing}, doi = {10.1088/1478-3975/13/3/035003}, }